README.md
In LiNk-NY/curatedTCGAManu: Presents example analyses using curatedTCGAData, MultiAssayExperiment, and TCGAutils
curatedTCGAManu
Multi-omic integration of public oncology databases in Bioconductor

This repository contains scripts and datasets for the curatedTCGAData +
cBioPortalData manuscript.
Overview
We provide several examples to demonstrate the powerful and flexible
analysis environment provided. These analyses, previously only
achievable through a significant investment of time and bioinformatic
training, become straight- forward analysis exercises provided as
vignettes.
Key Points
-
Key objective: To provide flexible, integrated multi-omic
representations of public oncology databases in R/Bioconductor with
grealy reduced data management overhead.
-
Knowledge generated: Our Bioconductor software packages provide a
novel approach to lower barriers to analysis and tool development
for the TCGA and cBioPortal databases.
-
Relevance: Our tools provide flexible, programmatic analysis of
hundreds of fully integrated multi’omic oncology datasets within an
ecosystem of multi-omic analysis tools.
Key Packages
- MultiAssayExperiment
- curatedTCGAData
- TCGAutils
- cBioPortalData
- EnrichmentBrowser
- GSEABenchmarkeR
- RTCGAToolbox
Installation
Until the release of Bioconductor 3.11 (scheduled for April 28, 2020),
it is strongly recommended to use the devel version of Bioconductor.
That version can be installed the traditional
way or by using
the Docker container.
Additionally until the release of Bioconductor 3.11, cBioPortalData
must be installed from GitHub as shown in the following code chunk which
installs all necessary packages either directly or as dependencies. Note
that this code chunk is not evaluated, because installation only needs
to be performed once.
BiocManager::install(c("cBioPortalData", "LiNk-NY/curatedTCGAManu"))
Vignette Build
Because of the size of the data, it is recommended that the vignettes be
built individually. If using RStudio, the user can simply open the
vignette and press the knit button. Otherwise, the package can be
built completely with vignettes by doing:
R CMD build curatedTCGAManu
in the command line or
BiocManager::install("Link-NY/curatedTCGAManu", build_vignettes = TRUE)
in R.
Loading packages
library(curatedTCGAData)
library(cBioPortalData)
library(TCGAutils)
library(GenomicDataCommons)
library(rtracklayer)
Note. For clarity, we include library commands within the
supplemental code chunks.
Figures
Ease-of-use schematic
Figure 1

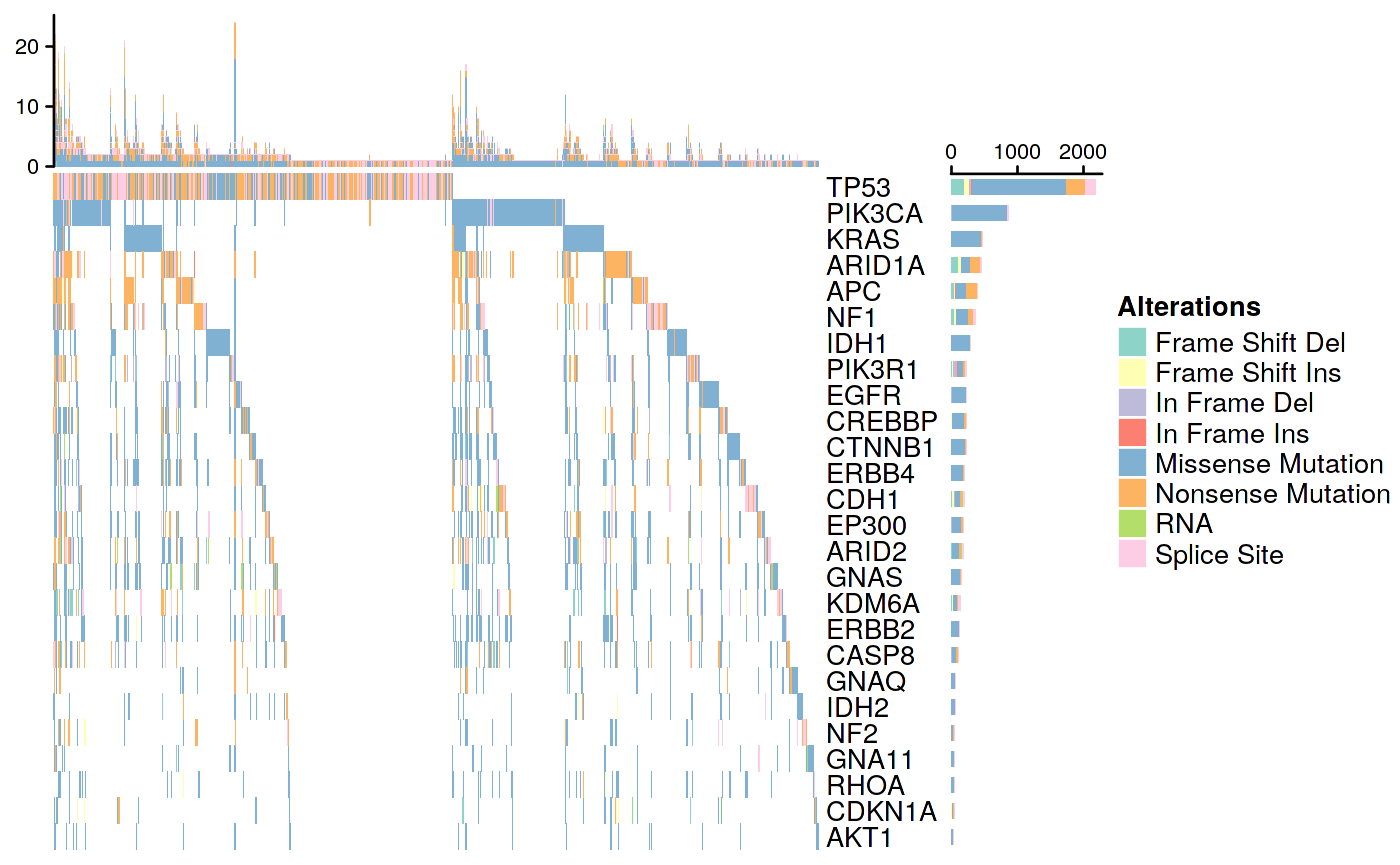
Pan-Cancer OncoPrint Plot
Figure 2
Differential Expression and GSEA PanCan
Figure 3
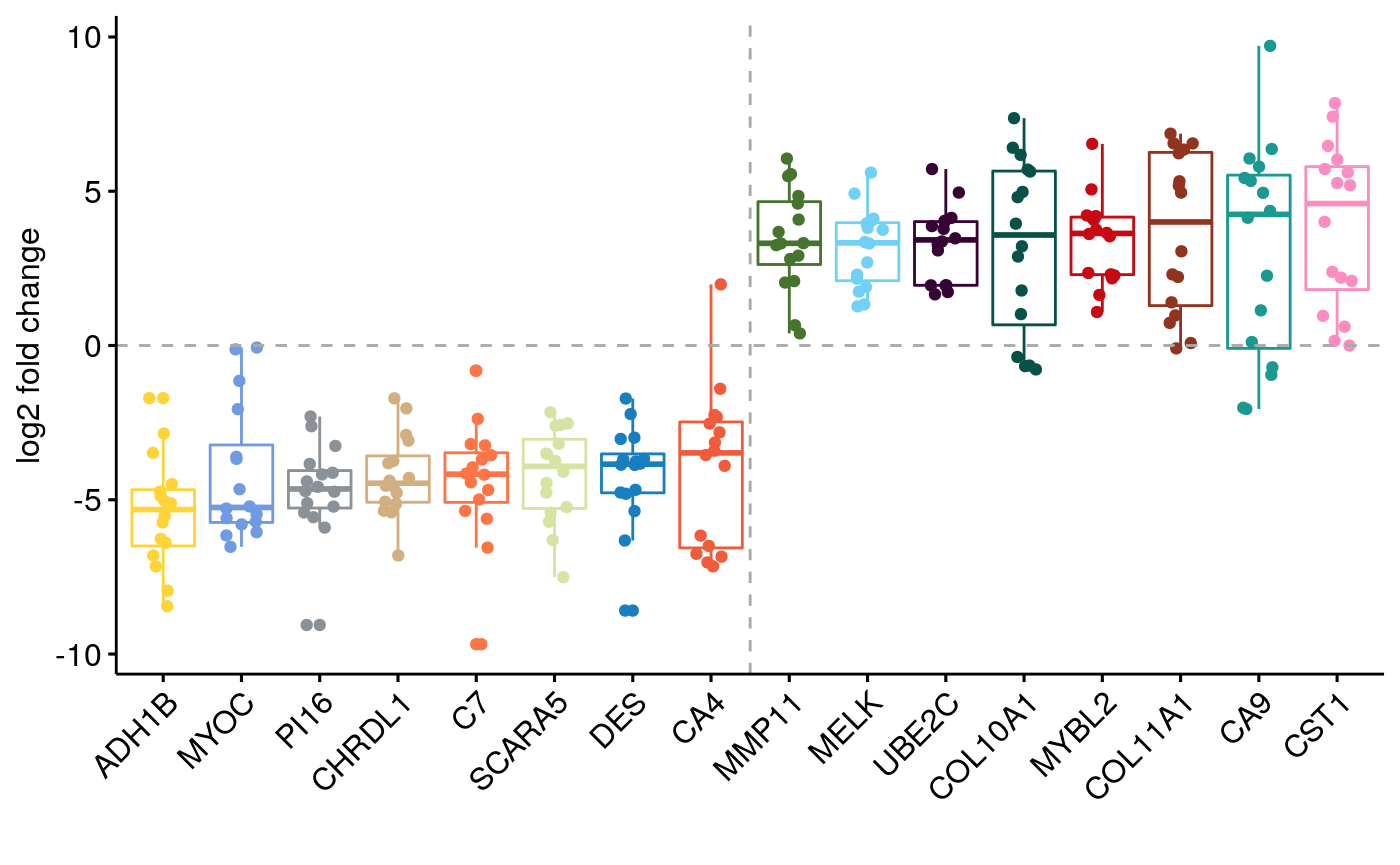
Figure 4

Example Multi-omic Analyses
Figure 5
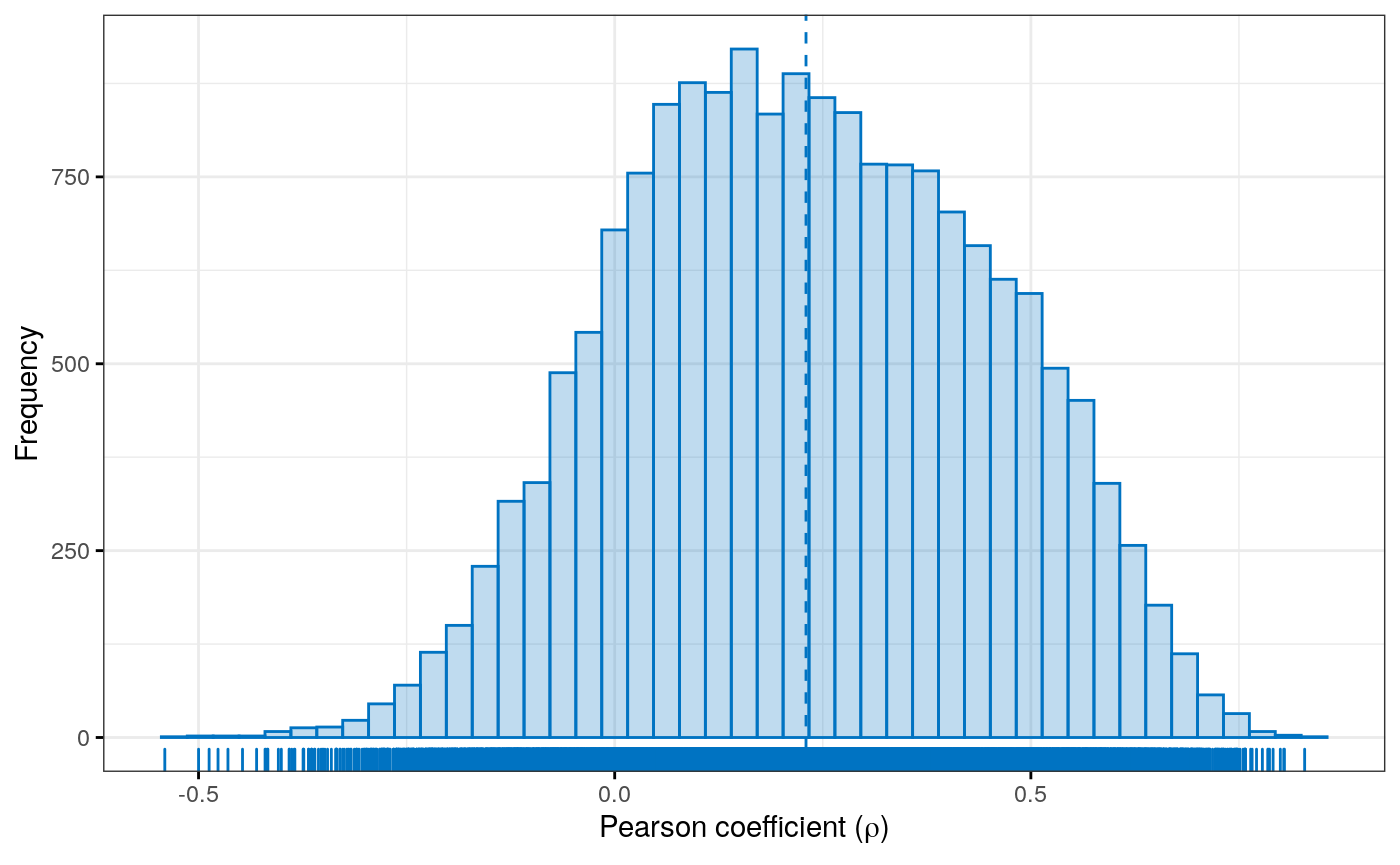
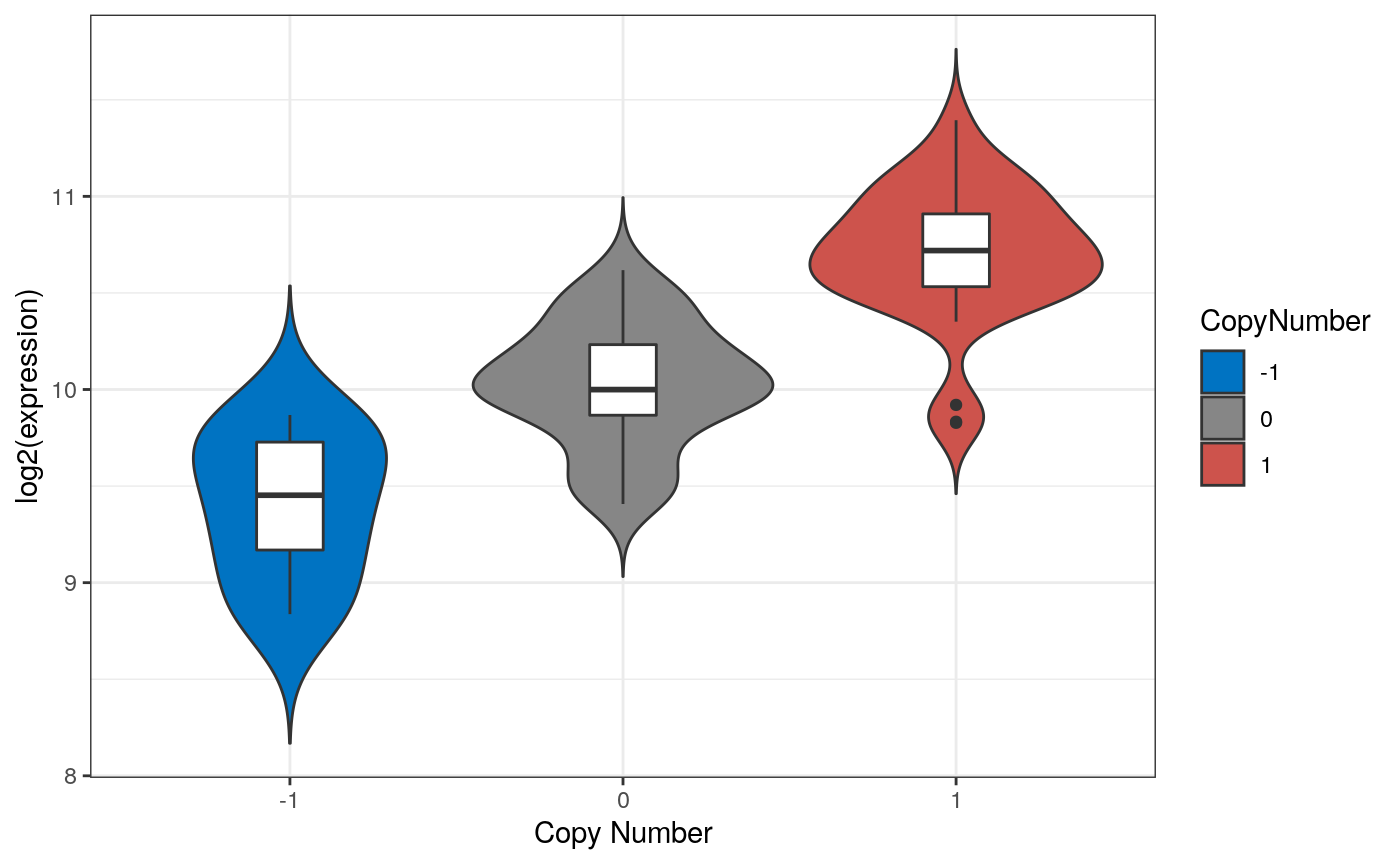
Figure 6
Supplement Reference
Figure S1
Data provenance and package network
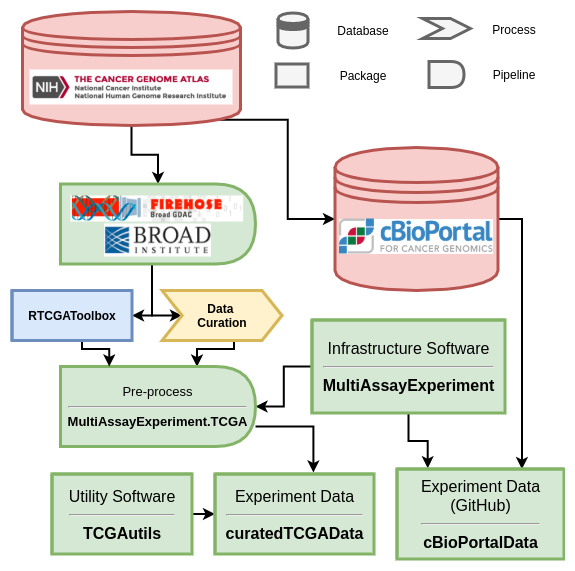
Figure S2A
Example code for installing and downloading TCGA data using curatedTCGAData
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
if (!requireNamespace("curatedTCGAData", quietly = TRUE))
BiocManager::install("curatedTCGAData")
## Glioblastoma Multiforme (GBM)
library(curatedTCGAData)
curatedTCGAData(diseaseCode = "GBM", assays = "RNA*", dry.run = FALSE)
#> A MultiAssayExperiment object of 1 listed
#> experiment with a user-defined name and respective class.
#> Containing an ExperimentList class object of length 1:
#> [1] GBM_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 166 columns
#> Functionality:
#> experiments() - obtain the ExperimentList instance
#> colData() - the primary/phenotype DataFrame
#> sampleMap() - the sample coordination DataFrame
#> `$`, `[`, `[[` - extract colData columns, subset, or experiment
#> *Format() - convert into a long or wide DataFrame
#> assays() - convert ExperimentList to a SimpleList of matrices
#> exportClass() - save all data to files
Figure S2B
Example cBioPortalData code for downloading and exporting TCGA data from cBioPortal.org and via the cBioPortal API
## installation
if (!requireNamespace("cBioPortalData", quietly = TRUE))
BiocManager::install("cBioPortalData")
library(cBioPortalData)
## https://cbioportal.org/datasets (Bulk data method)
gbm <- cBioDataPack("gbm_tcga")
## https://cBioPortal.org/api (API method)
cBio <- cBioPortal()
## use exportClass() with the result to save data files
tcga_gbm <- cBioPortalData(cBio, studyId = "gbm_tcga", genePanelId = "IMPACT341")
tcga_gbm
#> A MultiAssayExperiment object of 16 listed
#> experiments with user-defined names and respective classes.
#> Containing an ExperimentList class object of length 16:
#> [1] gbm_tcga_rppa: SummarizedExperiment with 67 rows and 244 columns
#> [2] gbm_tcga_rppa_Zscores: SummarizedExperiment with 67 rows and 244 columns
#> [3] gbm_tcga_gistic: SummarizedExperiment with 339 rows and 577 columns
#> [4] gbm_tcga_mrna_U133: SummarizedExperiment with 311 rows and 528 columns
#> [5] gbm_tcga_mrna_U133_Zscores: SummarizedExperiment with 308 rows and 528 columns
#> [6] gbm_tcga_mrna: SummarizedExperiment with 334 rows and 401 columns
#> [7] gbm_tcga_mrna_median_Zscores: SummarizedExperiment with 329 rows and 401 columns
#> [8] gbm_tcga_rna_seq_v2_mrna: SummarizedExperiment with 341 rows and 166 columns
#> [9] gbm_tcga_rna_seq_v2_mrna_median_Zscores: SummarizedExperiment with 333 rows and 166 columns
#> [10] gbm_tcga_linear_CNA: SummarizedExperiment with 339 rows and 577 columns
#> [11] gbm_tcga_methylation_hm27: SummarizedExperiment with 282 rows and 285 columns
#> [12] gbm_tcga_methylation_hm450: SummarizedExperiment with 282 rows and 153 columns
#> [13] gbm_tcga_mutations: RangedSummarizedExperiment with 810 rows and 271 columns
#> [14] gbm_tcga_rna_seq_v2_mrna_median_all_sample_Zscores: SummarizedExperiment with 340 rows and 166 columns
#> [15] gbm_tcga_mrna_median_all_sample_Zscores: SummarizedExperiment with 304 rows and 401 columns
#> [16] gbm_tcga_mrna_U133_all_sample_Zscores: SummarizedExperiment with 311 rows and 528 columns
#> Functionality:
#> experiments() - obtain the ExperimentList instance
#> colData() - the primary/phenotype DataFrame
#> sampleMap() - the sample coordination DataFrame
#> `$`, `[`, `[[` - extract colData columns, subset, or experiment
#> *Format() - convert into a long or wide DataFrame
#> assays() - convert ExperimentList to a SimpleList of matrices
#> exportClass() - save all data to files
exportClass(tcga_gbm, dir = tempdir(), fmt = "csv")
#> [1] "/tmp/Rtmp1EL92I/tcga_gbm_META_1.csv"
#> [2] "/tmp/Rtmp1EL92I/tcga_gbm_META_2.csv"
#> [3] "/tmp/Rtmp1EL92I/tcga_gbm_META_3.csv"
#> [4] "/tmp/Rtmp1EL92I/tcga_gbm_META_4.csv"
#> [5] "/tmp/Rtmp1EL92I/tcga_gbm_META_5.csv"
#> [6] "/tmp/Rtmp1EL92I/tcga_gbm_META_6.csv"
#> [7] "/tmp/Rtmp1EL92I/tcga_gbm_META_7.csv"
#> [8] "/tmp/Rtmp1EL92I/tcga_gbm_META_8.csv"
#> [9] "/tmp/Rtmp1EL92I/tcga_gbm_META_9.csv"
#> [10] "/tmp/Rtmp1EL92I/tcga_gbm_META_10.csv"
#> [11] "/tmp/Rtmp1EL92I/tcga_gbm_META_11.csv"
#> [12] "/tmp/Rtmp1EL92I/tcga_gbm_META_12.csv"
#> [13] "/tmp/Rtmp1EL92I/tcga_gbm_META_13.csv"
#> [14] "/tmp/Rtmp1EL92I/tcga_gbm_META_14.csv"
#> [15] "/tmp/Rtmp1EL92I/tcga_gbm_META_15.csv"
#> [16] "/tmp/Rtmp1EL92I/tcga_gbm_META_16.csv"
#> [17] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_rppa.csv"
#> [18] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_rppa_Zscores.csv"
#> [19] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_gistic.csv"
#> [20] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_mrna_U133.csv"
#> [21] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_mrna_U133_Zscores.csv"
#> [22] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_mrna.csv"
#> [23] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_mrna_median_Zscores.csv"
#> [24] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_rna_seq_v2_mrna.csv"
#> [25] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_rna_seq_v2_mrna_median_Zscores.csv"
#> [26] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_linear_CNA.csv"
#> [27] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_methylation_hm27.csv"
#> [28] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_methylation_hm450.csv"
#> [29] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_mutations.csv"
#> [30] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_rna_seq_v2_mrna_median_all_sample_Zscores.csv"
#> [31] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_mrna_median_all_sample_Zscores.csv"
#> [32] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_mrna_U133_all_sample_Zscores.csv"
#> [33] "/tmp/Rtmp1EL92I/tcga_gbm_colData.csv"
#> [34] "/tmp/Rtmp1EL92I/tcga_gbm_sampleMap.csv"
Figure S2C
Example hg19 to hg38 liftover procedure using Bioconductor tools
liftchain <- "http://hgdownload.cse.ucsc.edu/goldenpath/hg19/liftOver/hg19ToHg38.over.chain.gz"
cloc38 <- file.path(tempdir(), gsub("\\.gz", "", basename(liftchain)))
dfile <- tempfile(fileext = ".gz")
download.file(liftchain, dfile)
R.utils::gunzip(dfile, destname = cloc38, remove = FALSE)
library(rtracklayer)
chain38 <- suppressMessages( import.chain(cloc38) )
## Run bulk data download (from S2B) to create gbm object
if (!exists("gbm")) gbm <- cBioPortalData::cBioDataPack("gbm_tcga")
mutations <- gbm[["mutations_extended"]]
seqlevelsStyle(mutations) <- "UCSC"
ranges38 <- liftOver(rowRanges(mutations), chain38)
Figure S3
Example code for downloading data via GenomicDataCommons and loading with TCGAutils
library(TCGAutils)
library(GenomicDataCommons)
## GenomicDataCommons
query <- files(legacy = TRUE) %>%
filter( ~ cases.project.project_id == "TCGA-COAD" &
data_category == "Gene expression" &
data_type == "Exon quantification" )
fileids <- manifest(query)$id[1:4]
exonfiles <- gdcdata(fileids, use_cached = FALSE)
## TCGAutils
makeGRangesListFromExonFiles(exonfiles, nrows = 4)
#> Parsed with column specification:
#> cols(
#> exon = col_character(),
#> raw_counts = col_double(),
#> median_length_normalized = col_double(),
#> RPKM = col_double()
#> )
#> Parsed with column specification:
#> cols(
#> exon = col_character(),
#> raw_counts = col_double(),
#> median_length_normalized = col_double(),
#> RPKM = col_double()
#> )
#> Parsed with column specification:
#> cols(
#> exon = col_character(),
#> raw_counts = col_double(),
#> median_length_normalized = col_double(),
#> RPKM = col_double()
#> )
#> Parsed with column specification:
#> cols(
#> exon = col_character(),
#> raw_counts = col_double(),
#> median_length_normalized = col_double(),
#> RPKM = col_double()
#> )
#> GRangesList object of length 4:
#> $`TCGA-5M-AAT4-01A-11R-A41B-07`
#> GRanges object with 4 ranges and 3 metadata columns:
#> seqnames ranges strand | raw_counts median_length_normalized
#> <Rle> <IRanges> <Rle> | <numeric> <numeric>
#> [1] chr1 11874-12227 + | 1 0.135977
#> [2] chr1 12595-12721 + | 2 0.547619
#> [3] chr1 12613-12721 + | 2 0.472222
#> [4] chr1 12646-12697 + | 1 0.529412
#> RPKM
#> <numeric>
#> [1] 0.0228442
#> [2] 0.1273517
#> [3] 0.1483822
#> [4] 0.1555160
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths
#>
#> $`TCGA-A6-6782-01A-11R-1839-07`
#> GRanges object with 4 ranges and 3 metadata columns:
#> seqnames ranges strand | raw_counts median_length_normalized
#> <Rle> <IRanges> <Rle> | <numeric> <numeric>
#> [1] chr1 11874-12227 + | 35 0.784702
#> [2] chr1 12595-12721 + | 9 0.873016
#> [3] chr1 12613-12721 + | 9 0.851852
#> [4] chr1 12646-12697 + | 8 0.843137
#> RPKM
#> <numeric>
#> [1] 0.691243
#> [2] 0.495456
#> [3] 0.577274
#> [4] 1.075605
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths
#>
#> $`TCGA-AA-3678-01A-01R-0905-07`
#> GRanges object with 4 ranges and 3 metadata columns:
#> seqnames ranges strand | raw_counts median_length_normalized
#> <Rle> <IRanges> <Rle> | <numeric> <numeric>
#> [1] chr1 11874-12227 + | 4 0.492918
#> [2] chr1 12595-12721 + | 2 0.341270
#> [3] chr1 12613-12721 + | 2 0.398148
#> [4] chr1 12646-12697 + | 2 0.372549
#> RPKM
#> <numeric>
#> [1] 0.322477
#> [2] 0.449436
#> [3] 0.523655
#> [4] 1.097661
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths
#>
#> $`TCGA-AA-3955-01A-02R-1022-07`
#> GRanges object with 4 ranges and 3 metadata columns:
#> seqnames ranges strand | raw_counts median_length_normalized
#> <Rle> <IRanges> <Rle> | <numeric> <numeric>
#> [1] chr1 11874-12227 + | 0 0
#> [2] chr1 12595-12721 + | 0 0
#> [3] chr1 12613-12721 + | 0 0
#> [4] chr1 12646-12697 + | 0 0
#> RPKM
#> <numeric>
#> [1] 0
#> [2] 0
#> [3] 0
#> [4] 0
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths
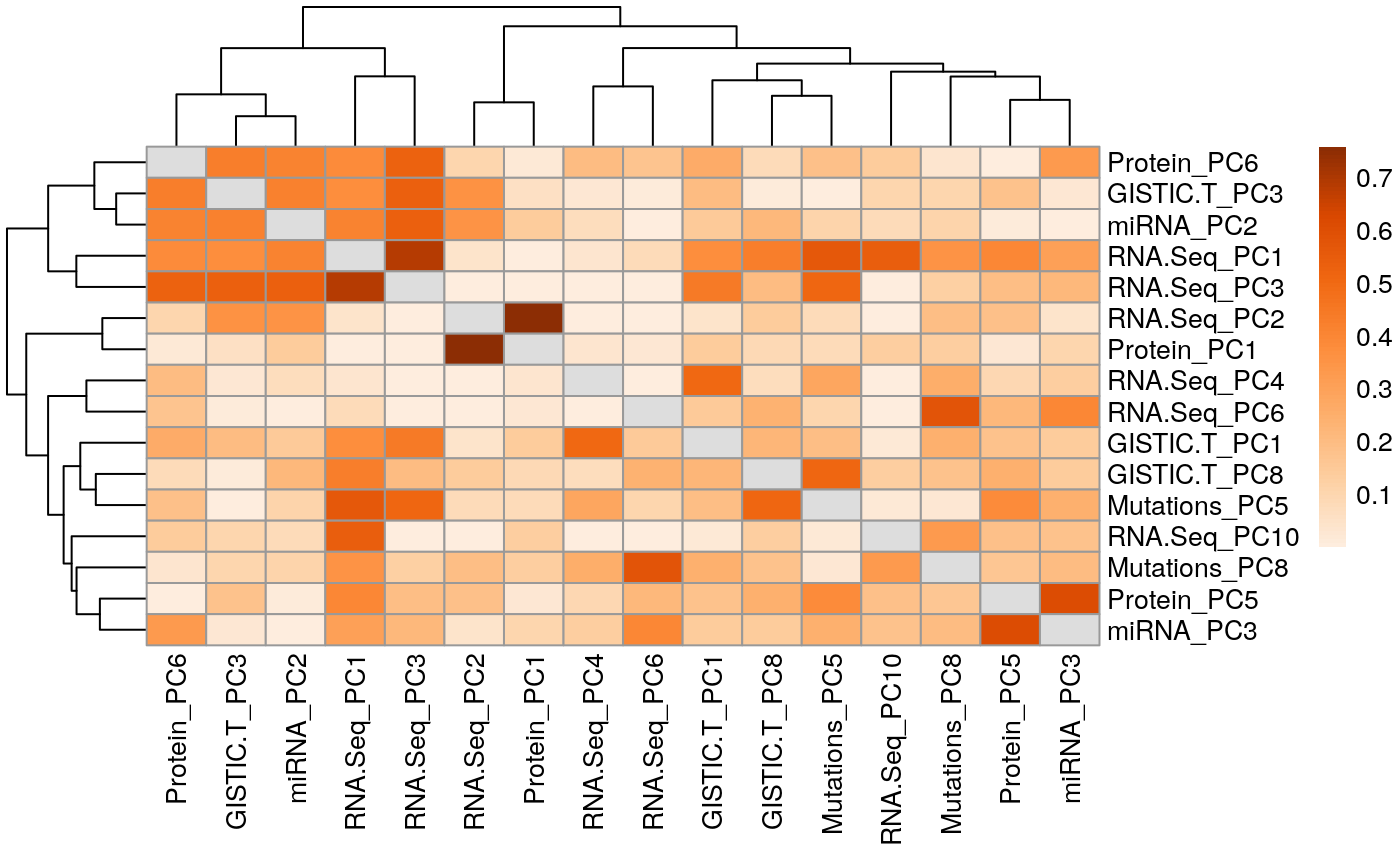
Figure S4
Correlated principal components across experimental assays in adrenocortical carcinoma (ACC)
LiNk-NY/curatedTCGAManu documentation built on July 22, 2020, 4:06 p.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
curatedTCGAManu
Multi-omic integration of public oncology databases in Bioconductor
![]()
This repository contains scripts and datasets for the curatedTCGAData + cBioPortalData manuscript.
Overview
We provide several examples to demonstrate the powerful and flexible analysis environment provided. These analyses, previously only achievable through a significant investment of time and bioinformatic training, become straight- forward analysis exercises provided as vignettes.
Key Points
-
Key objective: To provide flexible, integrated multi-omic representations of public oncology databases in R/Bioconductor with grealy reduced data management overhead.
-
Knowledge generated: Our Bioconductor software packages provide a novel approach to lower barriers to analysis and tool development for the TCGA and cBioPortal databases.
-
Relevance: Our tools provide flexible, programmatic analysis of hundreds of fully integrated multi’omic oncology datasets within an ecosystem of multi-omic analysis tools.
Key Packages
- MultiAssayExperiment
- curatedTCGAData
- TCGAutils
- cBioPortalData
- EnrichmentBrowser
- GSEABenchmarkeR
- RTCGAToolbox
Installation
Until the release of Bioconductor 3.11 (scheduled for April 28, 2020), it is strongly recommended to use the devel version of Bioconductor. That version can be installed the traditional way or by using the Docker container.
Additionally until the release of Bioconductor 3.11, cBioPortalData
must be installed from GitHub as shown in the following code chunk which
installs all necessary packages either directly or as dependencies. Note
that this code chunk is not evaluated, because installation only needs
to be performed once.
BiocManager::install(c("cBioPortalData", "LiNk-NY/curatedTCGAManu"))
Vignette Build
Because of the size of the data, it is recommended that the vignettes be
built individually. If using RStudio, the user can simply open the
vignette and press the knit button. Otherwise, the package can be
built completely with vignettes by doing:
R CMD build curatedTCGAManu
in the command line or
BiocManager::install("Link-NY/curatedTCGAManu", build_vignettes = TRUE)
in R.
Loading packages
library(curatedTCGAData)
library(cBioPortalData)
library(TCGAutils)
library(GenomicDataCommons)
library(rtracklayer)
Note. For clarity, we include library commands within the
supplemental code chunks.
Figures
Ease-of-use schematic
Figure 1
Pan-Cancer OncoPrint Plot
Figure 2
Differential Expression and GSEA PanCan
Figure 3
Figure 4
Example Multi-omic Analyses
Figure 5
Figure 6
Supplement Reference
Figure S1
Data provenance and package network
Figure S2A
Example code for installing and downloading TCGA data using curatedTCGAData
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
if (!requireNamespace("curatedTCGAData", quietly = TRUE))
BiocManager::install("curatedTCGAData")
## Glioblastoma Multiforme (GBM)
library(curatedTCGAData)
curatedTCGAData(diseaseCode = "GBM", assays = "RNA*", dry.run = FALSE)
#> A MultiAssayExperiment object of 1 listed
#> experiment with a user-defined name and respective class.
#> Containing an ExperimentList class object of length 1:
#> [1] GBM_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 166 columns
#> Functionality:
#> experiments() - obtain the ExperimentList instance
#> colData() - the primary/phenotype DataFrame
#> sampleMap() - the sample coordination DataFrame
#> `$`, `[`, `[[` - extract colData columns, subset, or experiment
#> *Format() - convert into a long or wide DataFrame
#> assays() - convert ExperimentList to a SimpleList of matrices
#> exportClass() - save all data to files
Figure S2B
Example cBioPortalData code for downloading and exporting TCGA data from cBioPortal.org and via the cBioPortal API
## installation
if (!requireNamespace("cBioPortalData", quietly = TRUE))
BiocManager::install("cBioPortalData")
library(cBioPortalData)
## https://cbioportal.org/datasets (Bulk data method)
gbm <- cBioDataPack("gbm_tcga")
## https://cBioPortal.org/api (API method)
cBio <- cBioPortal()
## use exportClass() with the result to save data files
tcga_gbm <- cBioPortalData(cBio, studyId = "gbm_tcga", genePanelId = "IMPACT341")
tcga_gbm
#> A MultiAssayExperiment object of 16 listed
#> experiments with user-defined names and respective classes.
#> Containing an ExperimentList class object of length 16:
#> [1] gbm_tcga_rppa: SummarizedExperiment with 67 rows and 244 columns
#> [2] gbm_tcga_rppa_Zscores: SummarizedExperiment with 67 rows and 244 columns
#> [3] gbm_tcga_gistic: SummarizedExperiment with 339 rows and 577 columns
#> [4] gbm_tcga_mrna_U133: SummarizedExperiment with 311 rows and 528 columns
#> [5] gbm_tcga_mrna_U133_Zscores: SummarizedExperiment with 308 rows and 528 columns
#> [6] gbm_tcga_mrna: SummarizedExperiment with 334 rows and 401 columns
#> [7] gbm_tcga_mrna_median_Zscores: SummarizedExperiment with 329 rows and 401 columns
#> [8] gbm_tcga_rna_seq_v2_mrna: SummarizedExperiment with 341 rows and 166 columns
#> [9] gbm_tcga_rna_seq_v2_mrna_median_Zscores: SummarizedExperiment with 333 rows and 166 columns
#> [10] gbm_tcga_linear_CNA: SummarizedExperiment with 339 rows and 577 columns
#> [11] gbm_tcga_methylation_hm27: SummarizedExperiment with 282 rows and 285 columns
#> [12] gbm_tcga_methylation_hm450: SummarizedExperiment with 282 rows and 153 columns
#> [13] gbm_tcga_mutations: RangedSummarizedExperiment with 810 rows and 271 columns
#> [14] gbm_tcga_rna_seq_v2_mrna_median_all_sample_Zscores: SummarizedExperiment with 340 rows and 166 columns
#> [15] gbm_tcga_mrna_median_all_sample_Zscores: SummarizedExperiment with 304 rows and 401 columns
#> [16] gbm_tcga_mrna_U133_all_sample_Zscores: SummarizedExperiment with 311 rows and 528 columns
#> Functionality:
#> experiments() - obtain the ExperimentList instance
#> colData() - the primary/phenotype DataFrame
#> sampleMap() - the sample coordination DataFrame
#> `$`, `[`, `[[` - extract colData columns, subset, or experiment
#> *Format() - convert into a long or wide DataFrame
#> assays() - convert ExperimentList to a SimpleList of matrices
#> exportClass() - save all data to files
exportClass(tcga_gbm, dir = tempdir(), fmt = "csv")
#> [1] "/tmp/Rtmp1EL92I/tcga_gbm_META_1.csv"
#> [2] "/tmp/Rtmp1EL92I/tcga_gbm_META_2.csv"
#> [3] "/tmp/Rtmp1EL92I/tcga_gbm_META_3.csv"
#> [4] "/tmp/Rtmp1EL92I/tcga_gbm_META_4.csv"
#> [5] "/tmp/Rtmp1EL92I/tcga_gbm_META_5.csv"
#> [6] "/tmp/Rtmp1EL92I/tcga_gbm_META_6.csv"
#> [7] "/tmp/Rtmp1EL92I/tcga_gbm_META_7.csv"
#> [8] "/tmp/Rtmp1EL92I/tcga_gbm_META_8.csv"
#> [9] "/tmp/Rtmp1EL92I/tcga_gbm_META_9.csv"
#> [10] "/tmp/Rtmp1EL92I/tcga_gbm_META_10.csv"
#> [11] "/tmp/Rtmp1EL92I/tcga_gbm_META_11.csv"
#> [12] "/tmp/Rtmp1EL92I/tcga_gbm_META_12.csv"
#> [13] "/tmp/Rtmp1EL92I/tcga_gbm_META_13.csv"
#> [14] "/tmp/Rtmp1EL92I/tcga_gbm_META_14.csv"
#> [15] "/tmp/Rtmp1EL92I/tcga_gbm_META_15.csv"
#> [16] "/tmp/Rtmp1EL92I/tcga_gbm_META_16.csv"
#> [17] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_rppa.csv"
#> [18] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_rppa_Zscores.csv"
#> [19] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_gistic.csv"
#> [20] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_mrna_U133.csv"
#> [21] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_mrna_U133_Zscores.csv"
#> [22] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_mrna.csv"
#> [23] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_mrna_median_Zscores.csv"
#> [24] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_rna_seq_v2_mrna.csv"
#> [25] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_rna_seq_v2_mrna_median_Zscores.csv"
#> [26] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_linear_CNA.csv"
#> [27] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_methylation_hm27.csv"
#> [28] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_methylation_hm450.csv"
#> [29] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_mutations.csv"
#> [30] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_rna_seq_v2_mrna_median_all_sample_Zscores.csv"
#> [31] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_mrna_median_all_sample_Zscores.csv"
#> [32] "/tmp/Rtmp1EL92I/tcga_gbm_gbm_tcga_mrna_U133_all_sample_Zscores.csv"
#> [33] "/tmp/Rtmp1EL92I/tcga_gbm_colData.csv"
#> [34] "/tmp/Rtmp1EL92I/tcga_gbm_sampleMap.csv"
Figure S2C
Example hg19 to hg38 liftover procedure using Bioconductor tools
liftchain <- "http://hgdownload.cse.ucsc.edu/goldenpath/hg19/liftOver/hg19ToHg38.over.chain.gz"
cloc38 <- file.path(tempdir(), gsub("\\.gz", "", basename(liftchain)))
dfile <- tempfile(fileext = ".gz")
download.file(liftchain, dfile)
R.utils::gunzip(dfile, destname = cloc38, remove = FALSE)
library(rtracklayer)
chain38 <- suppressMessages( import.chain(cloc38) )
## Run bulk data download (from S2B) to create gbm object
if (!exists("gbm")) gbm <- cBioPortalData::cBioDataPack("gbm_tcga")
mutations <- gbm[["mutations_extended"]]
seqlevelsStyle(mutations) <- "UCSC"
ranges38 <- liftOver(rowRanges(mutations), chain38)
Figure S3
Example code for downloading data via GenomicDataCommons and loading with TCGAutils
library(TCGAutils)
library(GenomicDataCommons)
## GenomicDataCommons
query <- files(legacy = TRUE) %>%
filter( ~ cases.project.project_id == "TCGA-COAD" &
data_category == "Gene expression" &
data_type == "Exon quantification" )
fileids <- manifest(query)$id[1:4]
exonfiles <- gdcdata(fileids, use_cached = FALSE)
## TCGAutils
makeGRangesListFromExonFiles(exonfiles, nrows = 4)
#> Parsed with column specification:
#> cols(
#> exon = col_character(),
#> raw_counts = col_double(),
#> median_length_normalized = col_double(),
#> RPKM = col_double()
#> )
#> Parsed with column specification:
#> cols(
#> exon = col_character(),
#> raw_counts = col_double(),
#> median_length_normalized = col_double(),
#> RPKM = col_double()
#> )
#> Parsed with column specification:
#> cols(
#> exon = col_character(),
#> raw_counts = col_double(),
#> median_length_normalized = col_double(),
#> RPKM = col_double()
#> )
#> Parsed with column specification:
#> cols(
#> exon = col_character(),
#> raw_counts = col_double(),
#> median_length_normalized = col_double(),
#> RPKM = col_double()
#> )
#> GRangesList object of length 4:
#> $`TCGA-5M-AAT4-01A-11R-A41B-07`
#> GRanges object with 4 ranges and 3 metadata columns:
#> seqnames ranges strand | raw_counts median_length_normalized
#> <Rle> <IRanges> <Rle> | <numeric> <numeric>
#> [1] chr1 11874-12227 + | 1 0.135977
#> [2] chr1 12595-12721 + | 2 0.547619
#> [3] chr1 12613-12721 + | 2 0.472222
#> [4] chr1 12646-12697 + | 1 0.529412
#> RPKM
#> <numeric>
#> [1] 0.0228442
#> [2] 0.1273517
#> [3] 0.1483822
#> [4] 0.1555160
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths
#>
#> $`TCGA-A6-6782-01A-11R-1839-07`
#> GRanges object with 4 ranges and 3 metadata columns:
#> seqnames ranges strand | raw_counts median_length_normalized
#> <Rle> <IRanges> <Rle> | <numeric> <numeric>
#> [1] chr1 11874-12227 + | 35 0.784702
#> [2] chr1 12595-12721 + | 9 0.873016
#> [3] chr1 12613-12721 + | 9 0.851852
#> [4] chr1 12646-12697 + | 8 0.843137
#> RPKM
#> <numeric>
#> [1] 0.691243
#> [2] 0.495456
#> [3] 0.577274
#> [4] 1.075605
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths
#>
#> $`TCGA-AA-3678-01A-01R-0905-07`
#> GRanges object with 4 ranges and 3 metadata columns:
#> seqnames ranges strand | raw_counts median_length_normalized
#> <Rle> <IRanges> <Rle> | <numeric> <numeric>
#> [1] chr1 11874-12227 + | 4 0.492918
#> [2] chr1 12595-12721 + | 2 0.341270
#> [3] chr1 12613-12721 + | 2 0.398148
#> [4] chr1 12646-12697 + | 2 0.372549
#> RPKM
#> <numeric>
#> [1] 0.322477
#> [2] 0.449436
#> [3] 0.523655
#> [4] 1.097661
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths
#>
#> $`TCGA-AA-3955-01A-02R-1022-07`
#> GRanges object with 4 ranges and 3 metadata columns:
#> seqnames ranges strand | raw_counts median_length_normalized
#> <Rle> <IRanges> <Rle> | <numeric> <numeric>
#> [1] chr1 11874-12227 + | 0 0
#> [2] chr1 12595-12721 + | 0 0
#> [3] chr1 12613-12721 + | 0 0
#> [4] chr1 12646-12697 + | 0 0
#> RPKM
#> <numeric>
#> [1] 0
#> [2] 0
#> [3] 0
#> [4] 0
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths
Figure S4
Correlated principal components across experimental assays in adrenocortical carcinoma (ACC)
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.