readme.md
In brandonsie/epitopefindr: Minimal Overlaps from BLAST Alignments
epitopefindr 




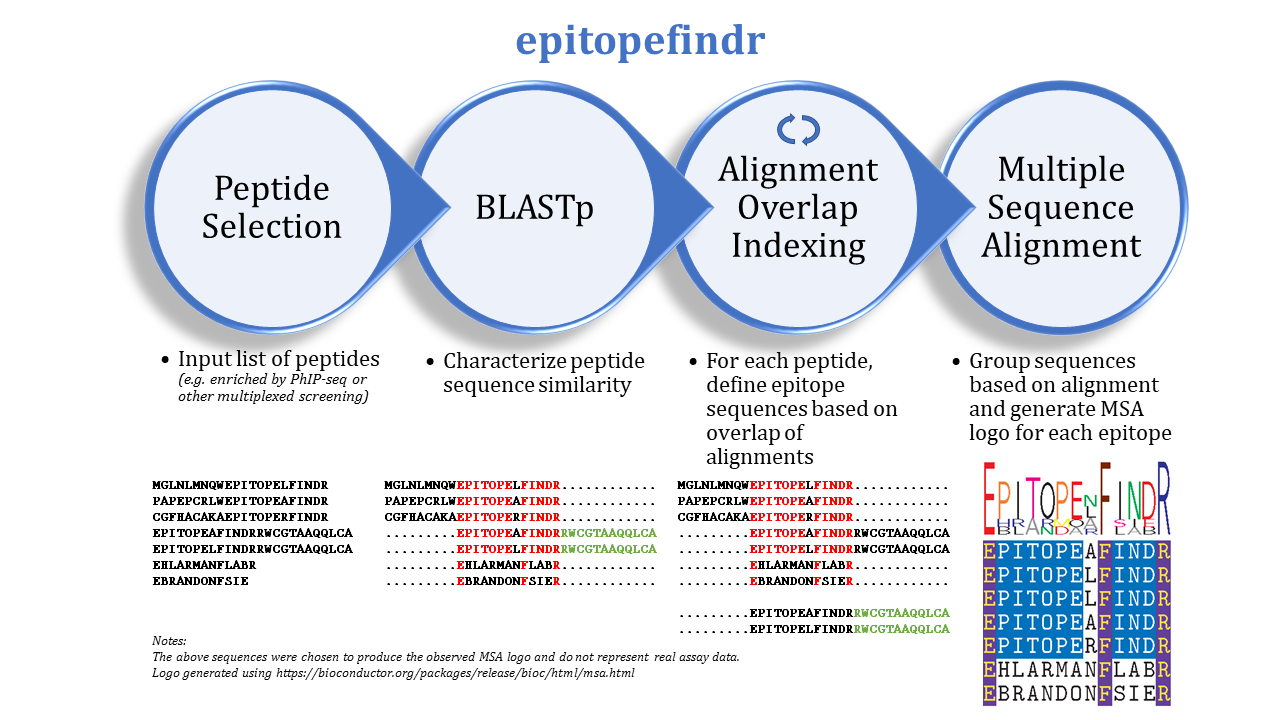
The purpose of this package is to describe the BLAST alignments among a set of peptide sequences by reporting the overlaps of each peptide's alignments to other peptides in the set. One can imagine inputting a list of peptides enriched by immunoprecipitation (e.g. by PhIP-seq) to identify corresponding epitopes.
epitopefindr takes a .fasta file listing peptide sequences of interest and calls BLASTp from within R to identify alignments among these peptides. Each peptide's alignments to other peptides are then simplified to the minimal number of "non overlapping" intervals of the index peptide that represent all alignments to other peptides reported by BLAST. (By default, each interval must be at least 7 amino acids long, and two intervals are considered NOT overlapping if they share 6 or fewer amino acids). After the minimal overlaps are identified for each peptide, these overlaps are gathered into aligning groups based on the initial BLAST. For each group, a multiple sequence alignment logo (motif) is generated to represent the collective sequence. Additionally, a spreadsheet is written to list the final trimmed amino acid sequences and some metadata.
Installation:
- Install R (version 3.5+).
- Install BLAST+ (version 2.7.1). (Note: we have observed some issues with more recent versions of BLAST+ and will monitor for bugfixes.)
- In R console, execute:
if (!requireNamespace("devtools")) install.packages("devtools")
devtools::install_github("brandonsie/epitopefindr")
library(epitopefindr)
Optional (Suggested) Additional Setup :
(These are not essential to epitopefindr, but are used to generate alignment logo PDFs from the alignment data, which can be valuable visualizations.)
1. Install a TeX distribution with pdflatex. (e.g. MiKTeX, Tex Live). (Optional; used to convert multiple sequence alignment TeX files to PDF.)
2. Install pdftk (version 2.02+). (Optional; used to merge individual PDFs into a single file.) If you are unable to install pdftk, but your system has the pdfunite command line utility, you can install the R package pdfuniter, which performs a similar function. With pdfuniter, run epfind with pdftk = FALSE, pdfunite = TRUE.
- as of epitopefindr version 1.1.30 (2020-09-20), pdftk = FALSE, pdfunite = TRUE is the default behavior. If your machine does not have the underlying pdfunite utility (e.g. macOS), try brew install poppler and then gem install pdfunite.
Debugging
- epitopefindr 1.1.29 (2020-05-30) updates the DESCRIPTION file to specify sources of Bioconductor and Github packages. If the above installation produces issues during certain package installations, try the following:
if (!requireNamespace("BiocManager")) install.packages("BiocManager")
BiocManager::install(c("Biostrings", "IRanges", "msa", "S4Vectors"))
# Install Github packages
if(!requireNamespace("devtools")) install.packages("devtools")
devtools::install_github("mhahsler/rBLAST")
devtools::install_github("brandonsie/pdfuniter")
devtools::install_github("brandonsie/epitopefindr")
Guide
- Prepare a list of your peptides of interest using one of the following two methods. Either of these can be fed as the first input parameter to
epfind.
- Make a FASTA file with peptide names and sequences.
- Make an
AAStringSet object of peptides (identifier + sequence) as described in the Biostrings documentation.
- To run a typical
epitopefindr pipeline, try calling epfind:
# Basic call
epfind(<path to .fasta>, <path to output dir>)
# Without pdflatex or pdftk
epfind(<path to .fasta>, <path to output dir>,
pdflatex = FALSE, pdftk = FALSE)
# More stringent e-value threshold
epfind(<path to .fasta>, <path to output dir>, e.thresh = 0.0001)
You can try running epfind() with some provided example data:
my_peptides <- epitopefindr::pairwise_viral_hits
epitopefindr::epfind(data = my_peptides, output.dir = "my_epf_1/")
A brief summary of the functions called by epfind:
* pbCycleBLAST cycles through each input peptide to find the overlap of its alignment with other peptides from the input. Nested within a call to pbCycleBLAST are calls to epitopeBLAST, indexEpitopes.
* trimEpitopes performs a second pass through the identified sequences to tidy alignments.
* indexGroups collects trimmed sequences into aligning groups
* groupMSA creates a multiple sequence alignment motif logo for each group
* outputTable creates a spreadsheet summarizing identified sequences and epitope groups
For more information, please visit:
brandonsie/epitopefindr documentation built on Oct. 31, 2021, 5:20 p.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
epitopefindr
![]()
![]()
The purpose of this package is to describe the BLAST alignments among a set of peptide sequences by reporting the overlaps of each peptide's alignments to other peptides in the set. One can imagine inputting a list of peptides enriched by immunoprecipitation (e.g. by PhIP-seq) to identify corresponding epitopes.
epitopefindr takes a .fasta file listing peptide sequences of interest and calls BLASTp from within R to identify alignments among these peptides. Each peptide's alignments to other peptides are then simplified to the minimal number of "non overlapping" intervals of the index peptide that represent all alignments to other peptides reported by BLAST. (By default, each interval must be at least 7 amino acids long, and two intervals are considered NOT overlapping if they share 6 or fewer amino acids). After the minimal overlaps are identified for each peptide, these overlaps are gathered into aligning groups based on the initial BLAST. For each group, a multiple sequence alignment logo (motif) is generated to represent the collective sequence. Additionally, a spreadsheet is written to list the final trimmed amino acid sequences and some metadata.
Installation:
- Install R (version 3.5+).
- Install BLAST+ (version 2.7.1). (Note: we have observed some issues with more recent versions of BLAST+ and will monitor for bugfixes.)
- In R console, execute:
if (!requireNamespace("devtools")) install.packages("devtools")
devtools::install_github("brandonsie/epitopefindr")
library(epitopefindr)
Optional (Suggested) Additional Setup :
(These are not essential to epitopefindr, but are used to generate alignment logo PDFs from the alignment data, which can be valuable visualizations.)
1. Install a TeX distribution with pdflatex. (e.g. MiKTeX, Tex Live). (Optional; used to convert multiple sequence alignment TeX files to PDF.)
2. Install pdftk (version 2.02+). (Optional; used to merge individual PDFs into a single file.) If you are unable to install pdftk, but your system has the pdfunite command line utility, you can install the R package pdfuniter, which performs a similar function. With pdfuniter, run epfind with pdftk = FALSE, pdfunite = TRUE.
- as of epitopefindr version 1.1.30 (2020-09-20), pdftk = FALSE, pdfunite = TRUE is the default behavior. If your machine does not have the underlying pdfunite utility (e.g. macOS), try brew install poppler and then gem install pdfunite.
Debugging
- epitopefindr 1.1.29 (2020-05-30) updates the DESCRIPTION file to specify sources of Bioconductor and Github packages. If the above installation produces issues during certain package installations, try the following:
if (!requireNamespace("BiocManager")) install.packages("BiocManager")
BiocManager::install(c("Biostrings", "IRanges", "msa", "S4Vectors"))
# Install Github packages
if(!requireNamespace("devtools")) install.packages("devtools")
devtools::install_github("mhahsler/rBLAST")
devtools::install_github("brandonsie/pdfuniter")
devtools::install_github("brandonsie/epitopefindr")
Guide
- Prepare a list of your peptides of interest using one of the following two methods. Either of these can be fed as the first input parameter to
epfind.- Make a FASTA file with peptide names and sequences.
- Make an
AAStringSetobject of peptides (identifier + sequence) as described in the Biostrings documentation.
- To run a typical
epitopefindrpipeline, try callingepfind:
# Basic call
epfind(<path to .fasta>, <path to output dir>)
# Without pdflatex or pdftk
epfind(<path to .fasta>, <path to output dir>,
pdflatex = FALSE, pdftk = FALSE)
# More stringent e-value threshold
epfind(<path to .fasta>, <path to output dir>, e.thresh = 0.0001)
You can try running epfind() with some provided example data:
my_peptides <- epitopefindr::pairwise_viral_hits
epitopefindr::epfind(data = my_peptides, output.dir = "my_epf_1/")
A brief summary of the functions called by epfind:
* pbCycleBLAST cycles through each input peptide to find the overlap of its alignment with other peptides from the input. Nested within a call to pbCycleBLAST are calls to epitopeBLAST, indexEpitopes.
* trimEpitopes performs a second pass through the identified sequences to tidy alignments.
* indexGroups collects trimmed sequences into aligning groups
* groupMSA creates a multiple sequence alignment motif logo for each group
* outputTable creates a spreadsheet summarizing identified sequences and epitope groups
For more information, please visit:
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.