README.md
In mararie/SAMBAR: Subtyping Agglomerated Mutations By Annotation Relations
SAMBAR
Subtyping Agglomerated Mutations By Annotation Relations
This R package depends on CRAN packages vegan, stats, and utils.
The easiest way to install the R package SAMBAR is via the devtools package from CRAN:
install.packages("devtools")
library(devtools)
devtools::install_github("mararie/SAMBAR")
And then load the package using: library(SAMBAR).
As an example, we've added mutation data of Uterine Corpus Endometrial Carcinoma (UCEC) primary tumor samples, obtained from The Cancer Genome Atlas, to this package. Running subtypes <- sambar() will run SAMBAR with default settings on these data, using desparsification based on based on the MSigDb "Hallmark" gene sets. It will return a list of samples belonging to k=2-4 subtypes.
More information on the method
SAMBAR, or Subtyping Agglomerated Mutations By Annotation Relations, is a method to identify subtypes based on somatic mutation data. SAMBAR was used to identify mutational subtypes in 23 cancer types from The Cancer Genome Atlas (Kuijjer ML, Paulson JN, Salzman P, Ding W, Quackenbush J, British Journal of Cancer (May 16, 2018), doi: 10.1038/s41416-018-0109-7, https://www.nature.com/articles/s41416-018-0109-7, BioRxiv, doi: https://doi.org/10.1101/228031).
SAMBAR's input is a matrix that includes the number of non-synonymous mutations in a sample 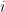 and gene
and gene 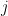 . SAMBAR first subsets these data to a set of 2,219 cancer-associated genes (optional) from the Catalogue Of Somatic Mutations In Cancer (COSMIC) and Östlund et al. (Network-based identification of novel cancer genes, 2010, Mol Cell Prot), or from a user-defined list. It then divides the number of non-synonymous mutations by the gene's length
. SAMBAR first subsets these data to a set of 2,219 cancer-associated genes (optional) from the Catalogue Of Somatic Mutations In Cancer (COSMIC) and Östlund et al. (Network-based identification of novel cancer genes, 2010, Mol Cell Prot), or from a user-defined list. It then divides the number of non-synonymous mutations by the gene's length  , defined as the number of non-overlapping exonic base pairs of a gene. For each sample, SAMBAR then calculates the overall cancer-associated mutation rate by summing mutation scores in all cancer-associated genes
, defined as the number of non-overlapping exonic base pairs of a gene. For each sample, SAMBAR then calculates the overall cancer-associated mutation rate by summing mutation scores in all cancer-associated genes 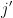 . It removes samples for which the mutation rate is zero and divides the mutation scores the remaining samples by the sample's mutation rate, resulting in a matrix of mutation rate-adjusted scores
. It removes samples for which the mutation rate is zero and divides the mutation scores the remaining samples by the sample's mutation rate, resulting in a matrix of mutation rate-adjusted scores 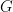 :
:
}) .
.
The next step in SAMBAR is de-sparsification of these gene mutation scores (agglomerated mutations) into pathway mutation (annotation relation) scores. SAMBAR converts a (user-defined) gene signature (.gmt format) into a binary matrix 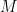 , with information of whether a gene belongs to a pathway
, with information of whether a gene belongs to a pathway  . It then calculates pathway mutation scores
. It then calculates pathway mutation scores  by correcting the sum of mutation scores of all genes in a pathway for the number of pathways
by correcting the sum of mutation scores of all genes in a pathway for the number of pathways 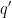 a gene belongs to, and for the number of cancer-associated genes present in that pathway:
a gene belongs to, and for the number of cancer-associated genes present in that pathway:
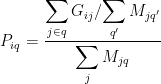 .
.
Finally, SAMBAR uses binomial distance to cluster the pathway mutation scores. The cluster dendrogram is then divided into k groups (or a range of k groups), and the cluster assignments are returned in a list.
mararie/SAMBAR documentation built on Nov. 23, 2019, 12:36 a.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
SAMBAR
Subtyping Agglomerated Mutations By Annotation Relations
This R package depends on CRAN packages vegan, stats, and utils.
The easiest way to install the R package SAMBAR is via the devtools package from CRAN:
install.packages("devtools")
library(devtools)
devtools::install_github("mararie/SAMBAR")
And then load the package using: library(SAMBAR).
As an example, we've added mutation data of Uterine Corpus Endometrial Carcinoma (UCEC) primary tumor samples, obtained from The Cancer Genome Atlas, to this package. Running subtypes <- sambar() will run SAMBAR with default settings on these data, using desparsification based on based on the MSigDb "Hallmark" gene sets. It will return a list of samples belonging to k=2-4 subtypes.
More information on the method
SAMBAR, or Subtyping Agglomerated Mutations By Annotation Relations, is a method to identify subtypes based on somatic mutation data. SAMBAR was used to identify mutational subtypes in 23 cancer types from The Cancer Genome Atlas (Kuijjer ML, Paulson JN, Salzman P, Ding W, Quackenbush J, British Journal of Cancer (May 16, 2018), doi: 10.1038/s41416-018-0109-7, https://www.nature.com/articles/s41416-018-0109-7, BioRxiv, doi: https://doi.org/10.1101/228031).
SAMBAR's input is a matrix that includes the number of non-synonymous mutations in a sample and gene
. SAMBAR first subsets these data to a set of 2,219 cancer-associated genes (optional) from the Catalogue Of Somatic Mutations In Cancer (COSMIC) and Östlund et al. (Network-based identification of novel cancer genes, 2010, Mol Cell Prot), or from a user-defined list. It then divides the number of non-synonymous mutations by the gene's length
, defined as the number of non-overlapping exonic base pairs of a gene. For each sample, SAMBAR then calculates the overall cancer-associated mutation rate by summing mutation scores in all cancer-associated genes
. It removes samples for which the mutation rate is zero and divides the mutation scores the remaining samples by the sample's mutation rate, resulting in a matrix of mutation rate-adjusted scores
:
.
The next step in SAMBAR is de-sparsification of these gene mutation scores (agglomerated mutations) into pathway mutation (annotation relation) scores. SAMBAR converts a (user-defined) gene signature (.gmt format) into a binary matrix , with information of whether a gene
belongs to a pathway
. It then calculates pathway mutation scores
by correcting the sum of mutation scores of all genes in a pathway for the number of pathways
a gene belongs to, and for the number of cancer-associated genes present in that pathway:
.
Finally, SAMBAR uses binomial distance to cluster the pathway mutation scores. The cluster dendrogram is then divided into k groups (or a range of k groups), and the cluster assignments are returned in a list.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.