Nothing
Plugins of BioInstaller Shiny Application
In BioInstaller: Integrator of Bioinformatics Resources
knitr::opts_chunk$set(echo = TRUE, screenshot.force = FALSE, comment = "#>", collapse = TRUE)
Introduction
Both the R files and plugin files (TOML format) are supported to extend and add new functions of BioInstaller Shiny application. Users can modifed the value of shiny_plugin_dir to use a custom plugins library. The plugins filename: shiny.easy_project.parameters.toml, shiny.maftools.parameters, etc. Now we use the "shiny.{toolname}.parameters.toml" rule to search the plugin file, or you can modifed the line 84 of global_bar.R to change the rule.
Demos
Plugin easy_project
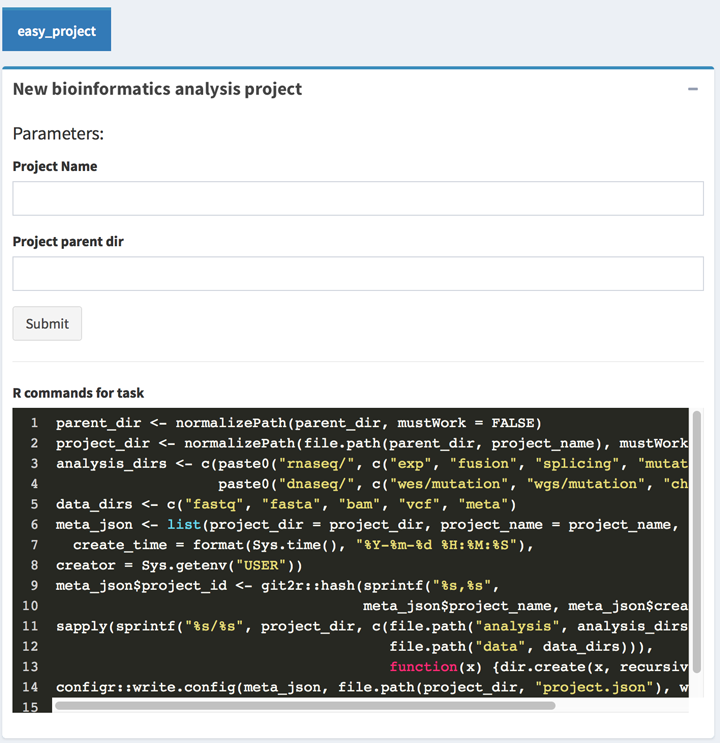
First level: ui, parameters
ui.sections defined the boxes, such as the new_proj.ui.sections.ui_basic defined the basic parameter of a box.
In the parameters, we could defined the section type, a simplest plugin of BioInstaller should only contains a input type box, and the box can input parameters and submit a long-time cost task.
rcmd_last contains the commands need to be ran in the workers.progressbar_message only for instant module plugins to shows the running boxrender_id is the prefix of the auto-generated boxes elementinput_ui_order defined the sections of input box, such as single_input and start_analysis
[easy_project.ui.sections]
order = ["new_proj"]
[easy_project.ui.sections.ui_basic]
new_proj = "title = 'New bioinformatics analysis project', status = 'primary', width = 12, collapsed = FALSE, collapsible = TRUE"
[easy_project.paramters.new_proj]
# For reading annovarR shiny APP easy_project tool input files
section_type = "input"
rcmd_last = """
parent_dir <- normalizePath(parent_dir, mustWork = FALSE)
project_dir <- normalizePath(file.path(parent_dir, project_name), mustWork = FALSE)
analysis_dirs <- c(paste0("rnaseq/", c("exp", "fusion", "splicing", "mutation")),
paste0("dnaseq/", c("wes/mutation", "wgs/mutation", "chip/peak")))
data_dirs <- c("fastq", "fasta", "bam", "vcf", "meta")
meta_json <- list(project_dir = project_dir, project_name = project_name,
create_time = format(Sys.time(), "%Y-%m-%d %H:%M:%S"),
creator = Sys.getenv("USER"))
meta_json$project_id <- git2r::hash(sprintf("%s,%s",
meta_json$project_name, meta_json$create_time))
sapply(sprintf("%s/%s", project_dir, c(file.path("analysis", analysis_dirs),
file.path("data", data_dirs))),
function(x) {dir.create(x, recursive = TRUE)})
configr::write.config(meta_json, file.path(project_dir, "project.json"), write.type = "json")
"""
progressbar_message = ""
render_id = "easy_project_new_proj"
#!!!!! input_ui_order required related section
input_ui_order = ["single_input", "start_analysis"]
[easy_project.paramters.new_proj.input.single_input]
title = "Parameters:"
title_control = "class = 'input-section-p'"
varname = ["project_name", "parent_dir"]
input_id = ["input_project_name", "input_parent_dir"]
type = ["shiny::textInput", "shiny::textInput"]
label = ["Project Name", "Project parent dir"]
[easy_project.paramters.new_proj.input.start_analysis]
input_id = "start_easy_project_analysis"
type = "shiny::actionButton"
label = "Submit"
In the parameters section, all variables in varname are automatically assigned to the element values of input_id, such as a text input box, selector or check box.
start_analysis section now is required for submitting a long-time cost task that this section should only need to change the plugin name (e.g. easy_project) only.
Plugin maftools
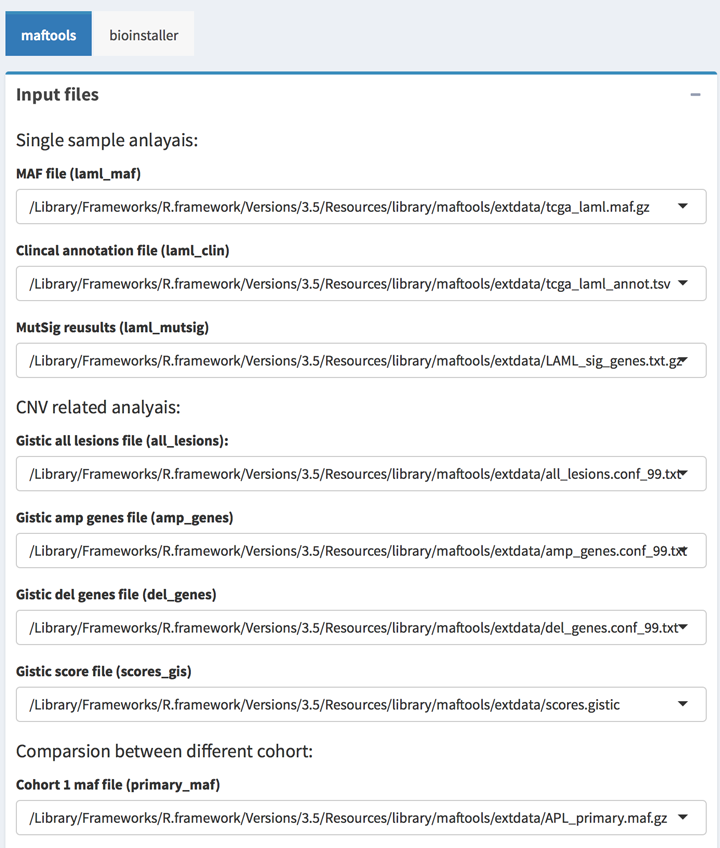

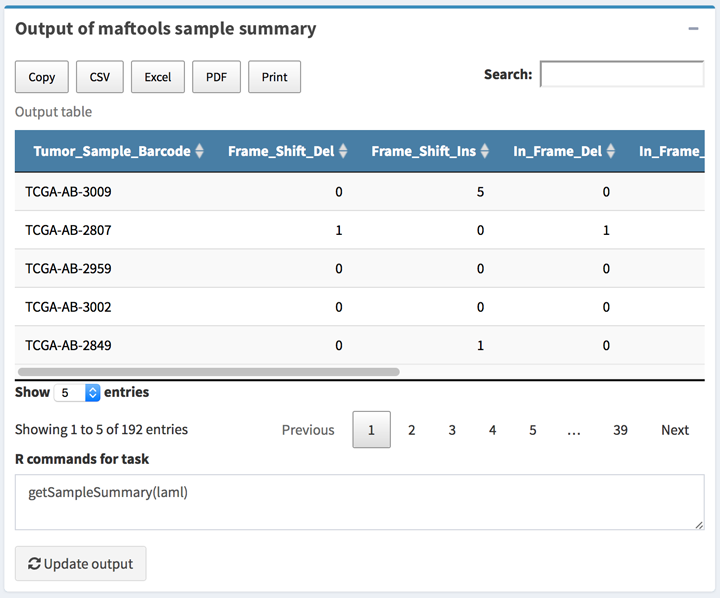
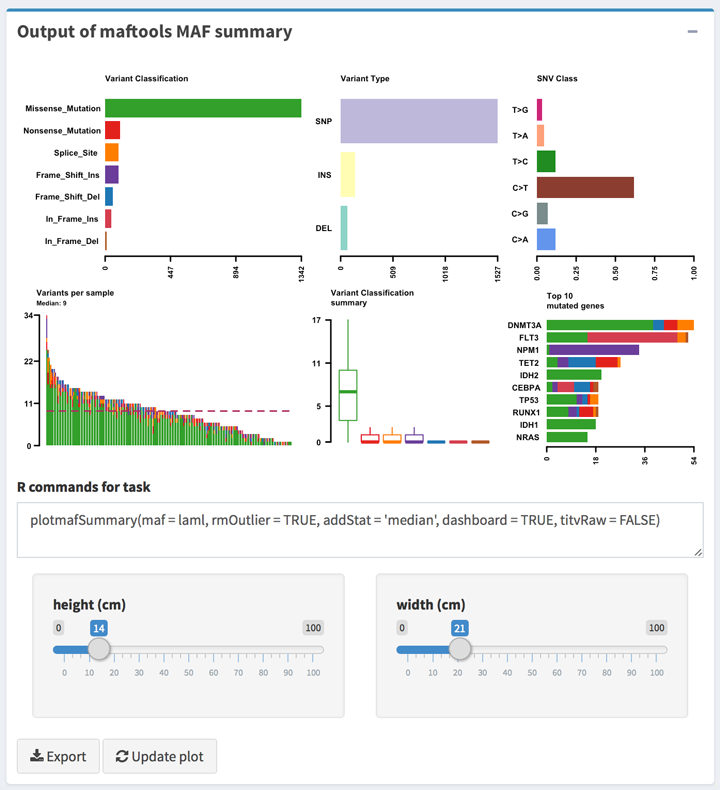
All output type boxes will get the instant output results including text, table, and plots. All result can be update when click the Update button on the box.
export_engine now support Cairo and link.
Cairo need a plot function in the rcmd_last, and a list such as list(src = "plot_output", alt = paste('Heatmap_3')), is required for export a plot through a file path.
[maftools.ui.sections]
order = ["readfiles", "getFields", "getSampleSummary", "plotmafSummary",
"oncoplot_default", "oncoplot_with_cnv", "oncoplot_advanced", "plotTiTv", "lollipopPlot2", "tcgaCompare", "plotVaf",
"somaticInteractions", "oncostrip", "plotEnrichmentResults", "plotClusters"]
order_bak = ["readfiles", "getFields", "getSampleSummary", "getGeneSummary", "getClinicalData", "plotmafSummary",
"oncoplot_default", "oncoplot_with_cnv", "oncoplot_advanced", "plotTiTv", "lollipopPlot", "lollipopPlot2", "tcgaCompare", "plotVaf",
"gisticChromPlot", "gisticBubblePlot", "gisticOncoPlot", "somaticInteractions", "oncostrip", "plotOncodrive",
"mafSurvival", "plotEnrichmentResults", "plotClusters"]
[maftools.ui.sections.ui_basic]
readfiles = "title = 'Input files', status = 'primary', width = 12, collapsed = FALSE, collapsible = TRUE"
getFields = "title = 'Output of maftools fields summary', status = 'primary', width = 12, collapsed = TRUE, collapsible = TRUE"
getSampleSummary = "title = 'Output of maftools sample summary', status = 'primary', width = 12, collapsed = TRUE, collapsible = TRUE"
getGeneSummary = "title = 'Output of maftools gene summary', status = 'primary', width = 12, collapsed = TRUE, collapsible = TRUE"
getClinicalData = "title = 'Output of maftools clinical data summary', status = 'primary', width = 12, collapsed = TRUE, collapsible = TRUE"
plotmafSummary = "title = 'Output of maftools MAF summary', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
oncoplot_default = "title = 'Output of maftools oncoplots', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
oncoplot_with_cnv = "title = 'Output of maftools oncoplots with copy number data', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
oncoplot_advanced = "title = 'Output of maftools oncoplots with advanced', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
plotTiTv = "title = 'Output of maftools transition and transversions', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
lollipopPlot = "title = 'Output of maftools Lollipop plots for amino acid changes', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
lollipopPlot2 = "title = 'Output of maftools Lollipop2 plots for amino acid changes', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
tcgaCompare = "title = 'Output of maftools comparing mutation load', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
plotVaf = "title = 'Output of maftools VAF boxplot', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
gisticChromPlot = "title = 'Output of maftools gistic genome plot', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
gisticBubblePlot = "title = 'Output of maftools gistic bubble plot', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
gisticOncoPlot = "title = 'Output of maftools gistic oncoplot plot', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
somaticInteractions = "title = 'Output of maftools somatic interactions', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
oncostrip = "title = 'Output of maftools somatic interactions (oncostrip)', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
plotOncodrive = "title = 'Output of maftools driver based on positional clustering', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
mafSurvival = "title = 'Output of maftools Survival', status = 'primary', width = 12, collapsed = TRUE, collapsible = TRUE"
plotEnrichmentResults = "title = 'Output of maftools Clinical enrichment analysis', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
plotClusters = "title = 'Output of maftools heterogeneity analysis', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE"
[maftools.paramters.readfiles]
# For reading annovarR shiny APP maftools tool input files
section_type = "input"
rcmd_last = """
laml = read.maf(maf = laml_maf, clinicalData = laml_clin)
laml.plus.gistic = read.maf(maf = laml_maf, gisticAllLesionsFile = all_lesions, gisticAmpGenesFile = amp_genes,
gisticDelGenesFile = del_genes, gisticScoresFile = scores_gis, isTCGA = is_tcga)
laml.gistic = readGistic(gisticAllLesionsFile = all_lesions, gisticAmpGenesFile = amp_genes, gisticDelGenesFile = del_genes, gisticScoresFile = scores_gis, isTCGA = is_tcga)
primary_maf = read.maf(maf = primary_maf)
relapse_maf = read.maf(maf = relapse_maf)
"""
progressbar_message = "Reading related MAF and other files."
render_id = "maftools_readfiles"
#!!!!! input_ui_order required related section
input_ui_order = ["single_input", "cnv_input", "comparsion_input", "other_params", "start_analysis"]
[maftools.paramters.readfiles.input.single_input]
title = "Single sample anlayais:"
title_control = "class = 'input-section-p'"
varname = ["laml_maf", "laml_clin", "laml_mutsig"]
input_id = ["input_maf", "input_clin", "input_mutsig"]
type = ["shiny::selectInput", "shiny::selectInput", "shiny::selectInput"]
label = ["MAF file (laml_maf)", "Clincal annotation file (laml_clin)", "MutSig reusults (laml_mutsig)"]
[maftools.paramters.readfiles.input.single_input.choices]
laml_maf = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(maf)', full.names = TRUE), featch_files(c('maf', 'maf.gz'))$file_path)}"
laml_clin = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(txt)|(tsv)', full.names = TRUE), featch_files(c('tsv', 'txt', 'txt.gz', 'tsv.gz'))$file_path)}"
laml_mutsig = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(txt)|(tsv)', full.names = TRUE), featch_files(c('tsv', 'txt', 'txt.gz', 'tsv.gz'))$file_path)}"
[maftools.paramters.readfiles.input.single_input.selected]
laml_maf = "@>@system.file('extdata', 'tcga_laml.maf.gz', package = 'maftools')@<@"
laml_clin = "@>@system.file('extdata', 'tcga_laml_annot.tsv', package = 'maftools')@<@"
laml_mutsig = "@>@system.file('extdata', 'LAML_sig_genes.txt.gz', package = 'maftools')@<@"
[maftools.paramters.readfiles.input.cnv_input]
title = "CNV related analyais:"
title_control = "class = 'input-section-p'"
varname = ["all_lesions", "amp_genes", "del_genes", "scores_gis"]
input_id = ["input_gistic_all_lesions", "input_gistic_amp", "input_gistic_del", "input_gistic_score"]
type = ["shiny::selectInput", "shiny::selectInput", "shiny::selectInput", "shiny::selectInput"]
label = ["Gistic all lesions file (all_lesions):", "Gistic amp genes file (amp_genes)",
"Gistic del genes file (del_genes)", "Gistic score file (scores_gis)"]
[maftools.paramters.readfiles.input.cnv_input.choices]
all_lesions = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(txt)|(tsv)', full.names = TRUE), featch_files(c('txt', 'txt.gz'))$file_path)}"
amp_genes = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(txt)|(tsv)', full.names = TRUE), featch_files(c('txt', 'txt.gz'))$file_path)}"
del_genes = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(txt)|(tsv)', full.names = TRUE), featch_files(c('txt', 'txt.gz'))$file_path)}"
scores_gis = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(gistic)', full.names = TRUE), featch_files(c('txt', 'txt.gz', 'gistic'))$file_path)}"
[maftools.paramters.readfiles.input.cnv_input.selected]
all_lesions = "@>@system.file('extdata', 'all_lesions.conf_99.txt', package = 'maftools')@<@"
amp_genes = "@>@system.file('extdata', 'amp_genes.conf_99.txt', package = 'maftools')@<@"
del_genes = "@>@system.file('extdata', 'del_genes.conf_99.txt', package = 'maftools')@<@"
scores_gis = "@>@system.file('extdata', 'scores.gistic', package = 'maftools')@<@"
[maftools.paramters.readfiles.input.comparsion_input]
title = "Comparsion between different cohort:"
title_control = "class = 'input-section-p'"
varname = ["primary_maf", "relapse_maf"]
input_id = ["input_primary_maf", "input_relapse_maf"]
type = ["shiny::selectInput", "shiny::selectInput"]
label = ["Cohort 1 maf file (primary_maf)", "Cohort 2 maf file (relapse_maf)"]
[maftools.paramters.readfiles.input.comparsion_input.choices]
primary_maf = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(maf)', full.names = TRUE), featch_files(c('maf', 'maf.gz'))$file_path)}"
relapse_maf = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(maf)', full.names = TRUE), featch_files(c('maf', 'maf.gz'))$file_path)}"
[maftools.paramters.readfiles.input.comparsion_input.selected]
primary_maf = "@>@system.file('extdata', 'APL_primary.maf.gz', package = 'maftools')@<@"
relapse_maf = "@>@system.file('extdata', 'APL_relapse.maf.gz', package = 'maftools')@<@"
[maftools.paramters.readfiles.input.other_params]
title = "Other paramters"
title_control = "class = 'input-section-p'"
varname = ["is_tcga"]
input_id = ["maftools_is_tcga"]
type = ["shiny::checkboxInput"]
label = ["Reading file: Maf file is TCGA format? (is_tcga is TRUE)",
"Command to read files."]
[maftools.paramters.readfiles.input.other_params.value]
is_tcga = true
[maftools.paramters.readfiles.input.start_analysis]
input_id = "start_maftools_analysis"
type = "shiny::actionButton"
label = "Run"
[maftools.paramters.getFields]
section_type = "output"
rcmd_last = "getFields(laml)"
render_type = "shiny::renderPrint"
render_id = "maftools_fields_summary"
output_type = "shiny::verbatimTextOutput"
progressbar_message = "Maftools getFields"
[maftools.paramters.getSampleSummary]
section_type = "output"
rcmd_last = "getSampleSummary(laml)"
render_type = "DT::renderDataTable"
render_id = "maftools_sample_summary"
output_type = "DT::dataTableOutput"
progressbar_message = "Maftools getSampleSummary"
[maftools.paramters.getGeneSummary]
section_type = "output"
rcmd_last = "getGeneSummary(laml)"
render_type = "DT::renderDataTable"
render_id = "maftools_gene_summary"
output_type = "DT::dataTableOutput"
progressbar_message = "Maftools getGeneSummary"
[maftools.paramters.getClinicalData]
section_type = "output"
render_type = "DT::renderDataTable"
render_id = "maftools_clinical_data"
output_type = "DT::dataTableOutput"
rcmd_last = "getClinicalData(laml)"
progressbar_message = "Maftools getClinicalData"
[maftools.paramters.plotmafSummary]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_maf_summary"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 14, units='cm', bg='transparent'"
rcmd_last = """plotmafSummary(maf = laml, rmOutlier = TRUE, addStat = 'median', dashboard = TRUE, titvRaw = FALSE)"""
progressbar_message = "Maftools plotmafSummary"
[maftools.paramters.oncoplot_default]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_oncoplots"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 14, units='cm', bg='transparent'"
rcmd_last = "oncoplot(maf = laml, top = 10, fontSize = 12)"
progressbar_message = "Maftools oncoplot_default"
[maftools.paramters.oncoplot_with_cnv]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_oncoplots_cnv"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 14, units='cm', bg='transparent'"
rcmd_last = "oncoplot(maf = laml.plus.gistic, top = 10, fontSize = 12)"
progressbar_message = "Maftools oncoplot with CNV"
[maftools.paramters.oncoplot_advanced]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_oncoplots_advanced"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 27, units='cm', bg='transparent'"
rcmd_preprocess = """
col = RColorBrewer::brewer.pal(n = 8, name = 'Paired')
names(col) = c('Frame_Shift_Del','Missense_Mutation', 'Nonsense_Mutation', 'Multi_Hit', 'Frame_Shift_Ins',
'In_Frame_Ins', 'Splice_Site', 'In_Frame_Del')
#Color coding for FAB classification; try getAnnotations(x = laml) to see available annotations.
fabcolors = RColorBrewer::brewer.pal(n = 8,name = 'Spectral')
names(fabcolors) = c("M0", "M1", "M2", "M3", "M4", "M5", "M6", "M7")
fabcolors = list(FAB_classification = fabcolors)
mutsigQval = 0.01
clinicalFeatures = "FAB_classification"
sortByAnnotation = TRUE
oncoplot_advanced_params <- list(maf = laml, colors = col, mutsig = laml_mutsig,
mutsigQval = mutsigQval, clinicalFeatures = clinicalFeatures,
sortByAnnotation = sortByAnnotation,
annotationColor = fabcolors)
"""
rcmd_last = """
do.call(oncoplot, oncoplot_advanced_params)
"""
progressbar_message = "Maftools oncoplot with advanced"
[maftools.paramters.plotTiTv]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_titv"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'"
rcmd_preprocess = "laml.titv = titv(maf = laml, plot = FALSE, useSyn = TRUE)"
rcmd_last = "plotTiTv(res = laml.titv)"
progressbar_message = "Maftools plotTiTv"
[maftools.paramters.lollipopPlot]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_lollipop"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'"
rcmd_last = """lollipopPlot(maf = laml, gene = 'DNMT3A', AACol = 'Protein_Change', showMutationRate = TRUE)"""
progressbar_message = "Maftools lollipopPlot"
[maftools.paramters.lollipopPlot2]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_lollipop2"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'"
rcmd_last = """lollipopPlot2(m1 = primary_maf, m2 = relapse_maf, gene = "PML",
AACol1 = "amino_acid_change", AACol2 = "amino_acid_change", m1_name = "Primary", m2_name = "Relapse")"""
progressbar_message = "Maftools lollipopPlot2"
[maftools.paramters.tcgaCompare]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_mutation_load"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'"
rcmd_last = "laml.mutload = tcgaCompare(maf = laml, cohortName = 'Example-LAML')"
progressbar_message = "Maftools tcgaCompare"
[maftools.paramters.plotVaf]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_vaf_box"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'"
rcmd_last = "plotVaf(maf = laml, vafCol = 'i_TumorVAF_WU')"
progressbar_message = "Maftools plotVaf"
[maftools.paramters.gisticChromPlot]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_gistic_genome"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'"
rcmd_last = """gisticChromPlot(gistic = laml.gistic, markBands = "all")"""
progressbar_message = "Maftools gisticChromPlot"
[maftools.paramters.gisticBubblePlot]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_gistic_bubble"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'"
rcmd_last = """gisticBubblePlot(gistic = laml.gistic)"""
progressbar_message = "Maftools gisticBubblePlot"
[maftools.paramters.gisticOncoPlot]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_gistic_oncoplot"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'"
rcmd_last = """gisticOncoPlot(gistic = laml.gistic, clinicalData = getClinicalData(x = laml),
clinicalFeatures = 'FAB_classification', sortByAnnotation = TRUE, top = 10)"""
progressbar_message = "Maftools gisticOncoPlot"
[maftools.paramters.somaticInteractions]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_somatic_inter"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'"
rcmd_last = """somaticInteractions(maf = laml, top = 25, pvalue = c(0.05, 0.1))"""
progressbar_message = "Maftools somaticInteractions"
[maftools.paramters.oncostrip]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_somatic_inter_oncostrip"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'"
rcmd_last = """oncostrip(maf = laml, genes = c('TP53', 'FLT3', 'RUNX1'))"""
progressbar_message = "Maftools oncostrip"
[maftools.paramters.plotOncodrive]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_oncodrive"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'"
rcmd_preprocess = """laml.sig = oncodrive(maf = laml, AACol = 'Protein_Change', minMut = 5, pvalMethod = 'zscore')"""
rcmd_last = """plotOncodrive(res = laml.sig, fdrCutOff = 0.1, useFraction = TRUE)"""
progressbar_message = "Maftools plotOncodrive"
[maftools.paramters.mafSurvival]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_survival"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'"
rcmd_last = """mafSurvival(maf = laml, genes = 'DNMT3A', time = 'days_to_last_followup',
Status = 'Overall_Survival_Status', isTCGA = TRUE)"""
progressbar_message = "Maftools mafSurvival"
[maftools.paramters.plotEnrichmentResults]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_clinical_enrichment"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'"
rcmd_preprocess = "fab.ce = clinicalEnrichment(maf = laml, clinicalFeature = 'FAB_classification')"
rcmd_last = """plotEnrichmentResults(enrich_res = fab.ce, pVal = 0.05)"""
progressbar_message = "Maftools plotEnrichmentResults"
[maftools.paramters.plotClusters]
section_type = "output"
render_type = "shiny::renderPlot"
render_id = "maftools_plot_clusters"
output_type = "shiny::plotOutput"
export_engine = "Cairo"
export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'"
rcmd_preprocess = "tcga.ab.2972.het = inferHeterogeneity(maf = laml, tsb = 'TCGA-AB-2972', vafCol = 'i_TumorVAF_WU')"
rcmd_last = "plotClusters(clusters = tcga.ab.2972.het)"
progressbar_message = "Maftools maftools_plot_clusters"
More demos
In another project annovarR, we developed several plugins files that have not been included in BioInstaller package, such as gvmap, clusterProfiler, ANNOVAR, CEMiTool, vcfanno, annovarR, etc. It is noted that we do not want to limite users to write R codes for extending the R shiny functions. But using the plugin files to simultaneously generate UI and server code is a good choice to simplify the steps for adding new functions in a Shiny application.
Try the BioInstaller package in your browser
Any scripts or data that you put into this service are public.
BioInstaller documentation built on May 1, 2019, 11:17 p.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
knitr::opts_chunk$set(echo = TRUE, screenshot.force = FALSE, comment = "#>", collapse = TRUE)
Introduction
Both the R files and plugin files (TOML format) are supported to extend and add new functions of BioInstaller Shiny application. Users can modifed the value of shiny_plugin_dir to use a custom plugins library. The plugins filename: shiny.easy_project.parameters.toml, shiny.maftools.parameters, etc. Now we use the "shiny.{toolname}.parameters.toml" rule to search the plugin file, or you can modifed the line 84 of global_bar.R to change the rule.
Demos
Plugin easy_project
First level: ui, parameters
ui.sectionsdefined the boxes, such as thenew_proj.ui.sections.ui_basicdefined the basic parameter of a box.
In the parameters, we could defined the section type, a simplest plugin of BioInstaller should only contains a input type box, and the box can input parameters and submit a long-time cost task.
rcmd_lastcontains the commands need to be ran in the workers.progressbar_messageonly forinstantmodule plugins to shows the running boxrender_idis the prefix of the auto-generated boxes elementinput_ui_orderdefined the sections ofinputbox, such assingle_inputandstart_analysis
[easy_project.ui.sections] order = ["new_proj"] [easy_project.ui.sections.ui_basic] new_proj = "title = 'New bioinformatics analysis project', status = 'primary', width = 12, collapsed = FALSE, collapsible = TRUE" [easy_project.paramters.new_proj] # For reading annovarR shiny APP easy_project tool input files section_type = "input" rcmd_last = """ parent_dir <- normalizePath(parent_dir, mustWork = FALSE) project_dir <- normalizePath(file.path(parent_dir, project_name), mustWork = FALSE) analysis_dirs <- c(paste0("rnaseq/", c("exp", "fusion", "splicing", "mutation")), paste0("dnaseq/", c("wes/mutation", "wgs/mutation", "chip/peak"))) data_dirs <- c("fastq", "fasta", "bam", "vcf", "meta") meta_json <- list(project_dir = project_dir, project_name = project_name, create_time = format(Sys.time(), "%Y-%m-%d %H:%M:%S"), creator = Sys.getenv("USER")) meta_json$project_id <- git2r::hash(sprintf("%s,%s", meta_json$project_name, meta_json$create_time)) sapply(sprintf("%s/%s", project_dir, c(file.path("analysis", analysis_dirs), file.path("data", data_dirs))), function(x) {dir.create(x, recursive = TRUE)}) configr::write.config(meta_json, file.path(project_dir, "project.json"), write.type = "json") """ progressbar_message = "" render_id = "easy_project_new_proj" #!!!!! input_ui_order required related section input_ui_order = ["single_input", "start_analysis"] [easy_project.paramters.new_proj.input.single_input] title = "Parameters:" title_control = "class = 'input-section-p'" varname = ["project_name", "parent_dir"] input_id = ["input_project_name", "input_parent_dir"] type = ["shiny::textInput", "shiny::textInput"] label = ["Project Name", "Project parent dir"] [easy_project.paramters.new_proj.input.start_analysis] input_id = "start_easy_project_analysis" type = "shiny::actionButton" label = "Submit"
In the parameters section, all variables in varname are automatically assigned to the element values of input_id, such as a text input box, selector or check box.
start_analysissection now is required for submitting a long-time cost task that this section should only need to change the plugin name (e.g. easy_project) only.
Plugin maftools
All output type boxes will get the instant output results including text, table, and plots. All result can be update when click the Update button on the box.
export_enginenow supportCairoandlink.
Cairo need a plot function in the rcmd_last, and a list such as list(src = "plot_output", alt = paste('Heatmap_3')), is required for export a plot through a file path.
[maftools.ui.sections] order = ["readfiles", "getFields", "getSampleSummary", "plotmafSummary", "oncoplot_default", "oncoplot_with_cnv", "oncoplot_advanced", "plotTiTv", "lollipopPlot2", "tcgaCompare", "plotVaf", "somaticInteractions", "oncostrip", "plotEnrichmentResults", "plotClusters"] order_bak = ["readfiles", "getFields", "getSampleSummary", "getGeneSummary", "getClinicalData", "plotmafSummary", "oncoplot_default", "oncoplot_with_cnv", "oncoplot_advanced", "plotTiTv", "lollipopPlot", "lollipopPlot2", "tcgaCompare", "plotVaf", "gisticChromPlot", "gisticBubblePlot", "gisticOncoPlot", "somaticInteractions", "oncostrip", "plotOncodrive", "mafSurvival", "plotEnrichmentResults", "plotClusters"] [maftools.ui.sections.ui_basic] readfiles = "title = 'Input files', status = 'primary', width = 12, collapsed = FALSE, collapsible = TRUE" getFields = "title = 'Output of maftools fields summary', status = 'primary', width = 12, collapsed = TRUE, collapsible = TRUE" getSampleSummary = "title = 'Output of maftools sample summary', status = 'primary', width = 12, collapsed = TRUE, collapsible = TRUE" getGeneSummary = "title = 'Output of maftools gene summary', status = 'primary', width = 12, collapsed = TRUE, collapsible = TRUE" getClinicalData = "title = 'Output of maftools clinical data summary', status = 'primary', width = 12, collapsed = TRUE, collapsible = TRUE" plotmafSummary = "title = 'Output of maftools MAF summary', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" oncoplot_default = "title = 'Output of maftools oncoplots', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" oncoplot_with_cnv = "title = 'Output of maftools oncoplots with copy number data', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" oncoplot_advanced = "title = 'Output of maftools oncoplots with advanced', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" plotTiTv = "title = 'Output of maftools transition and transversions', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" lollipopPlot = "title = 'Output of maftools Lollipop plots for amino acid changes', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" lollipopPlot2 = "title = 'Output of maftools Lollipop2 plots for amino acid changes', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" tcgaCompare = "title = 'Output of maftools comparing mutation load', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" plotVaf = "title = 'Output of maftools VAF boxplot', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" gisticChromPlot = "title = 'Output of maftools gistic genome plot', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" gisticBubblePlot = "title = 'Output of maftools gistic bubble plot', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" gisticOncoPlot = "title = 'Output of maftools gistic oncoplot plot', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" somaticInteractions = "title = 'Output of maftools somatic interactions', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" oncostrip = "title = 'Output of maftools somatic interactions (oncostrip)', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" plotOncodrive = "title = 'Output of maftools driver based on positional clustering', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" mafSurvival = "title = 'Output of maftools Survival', status = 'primary', width = 12, collapsed = TRUE, collapsible = TRUE" plotEnrichmentResults = "title = 'Output of maftools Clinical enrichment analysis', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" plotClusters = "title = 'Output of maftools heterogeneity analysis', status = 'primary', width = 12, collapsed = TRUE, height='auto', collapsible = TRUE" [maftools.paramters.readfiles] # For reading annovarR shiny APP maftools tool input files section_type = "input" rcmd_last = """ laml = read.maf(maf = laml_maf, clinicalData = laml_clin) laml.plus.gistic = read.maf(maf = laml_maf, gisticAllLesionsFile = all_lesions, gisticAmpGenesFile = amp_genes, gisticDelGenesFile = del_genes, gisticScoresFile = scores_gis, isTCGA = is_tcga) laml.gistic = readGistic(gisticAllLesionsFile = all_lesions, gisticAmpGenesFile = amp_genes, gisticDelGenesFile = del_genes, gisticScoresFile = scores_gis, isTCGA = is_tcga) primary_maf = read.maf(maf = primary_maf) relapse_maf = read.maf(maf = relapse_maf) """ progressbar_message = "Reading related MAF and other files." render_id = "maftools_readfiles" #!!!!! input_ui_order required related section input_ui_order = ["single_input", "cnv_input", "comparsion_input", "other_params", "start_analysis"] [maftools.paramters.readfiles.input.single_input] title = "Single sample anlayais:" title_control = "class = 'input-section-p'" varname = ["laml_maf", "laml_clin", "laml_mutsig"] input_id = ["input_maf", "input_clin", "input_mutsig"] type = ["shiny::selectInput", "shiny::selectInput", "shiny::selectInput"] label = ["MAF file (laml_maf)", "Clincal annotation file (laml_clin)", "MutSig reusults (laml_mutsig)"] [maftools.paramters.readfiles.input.single_input.choices] laml_maf = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(maf)', full.names = TRUE), featch_files(c('maf', 'maf.gz'))$file_path)}" laml_clin = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(txt)|(tsv)', full.names = TRUE), featch_files(c('tsv', 'txt', 'txt.gz', 'tsv.gz'))$file_path)}" laml_mutsig = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(txt)|(tsv)', full.names = TRUE), featch_files(c('tsv', 'txt', 'txt.gz', 'tsv.gz'))$file_path)}" [maftools.paramters.readfiles.input.single_input.selected] laml_maf = "@>@system.file('extdata', 'tcga_laml.maf.gz', package = 'maftools')@<@" laml_clin = "@>@system.file('extdata', 'tcga_laml_annot.tsv', package = 'maftools')@<@" laml_mutsig = "@>@system.file('extdata', 'LAML_sig_genes.txt.gz', package = 'maftools')@<@" [maftools.paramters.readfiles.input.cnv_input] title = "CNV related analyais:" title_control = "class = 'input-section-p'" varname = ["all_lesions", "amp_genes", "del_genes", "scores_gis"] input_id = ["input_gistic_all_lesions", "input_gistic_amp", "input_gistic_del", "input_gistic_score"] type = ["shiny::selectInput", "shiny::selectInput", "shiny::selectInput", "shiny::selectInput"] label = ["Gistic all lesions file (all_lesions):", "Gistic amp genes file (amp_genes)", "Gistic del genes file (del_genes)", "Gistic score file (scores_gis)"] [maftools.paramters.readfiles.input.cnv_input.choices] all_lesions = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(txt)|(tsv)', full.names = TRUE), featch_files(c('txt', 'txt.gz'))$file_path)}" amp_genes = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(txt)|(tsv)', full.names = TRUE), featch_files(c('txt', 'txt.gz'))$file_path)}" del_genes = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(txt)|(tsv)', full.names = TRUE), featch_files(c('txt', 'txt.gz'))$file_path)}" scores_gis = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(gistic)', full.names = TRUE), featch_files(c('txt', 'txt.gz', 'gistic'))$file_path)}" [maftools.paramters.readfiles.input.cnv_input.selected] all_lesions = "@>@system.file('extdata', 'all_lesions.conf_99.txt', package = 'maftools')@<@" amp_genes = "@>@system.file('extdata', 'amp_genes.conf_99.txt', package = 'maftools')@<@" del_genes = "@>@system.file('extdata', 'del_genes.conf_99.txt', package = 'maftools')@<@" scores_gis = "@>@system.file('extdata', 'scores.gistic', package = 'maftools')@<@" [maftools.paramters.readfiles.input.comparsion_input] title = "Comparsion between different cohort:" title_control = "class = 'input-section-p'" varname = ["primary_maf", "relapse_maf"] input_id = ["input_primary_maf", "input_relapse_maf"] type = ["shiny::selectInput", "shiny::selectInput"] label = ["Cohort 1 maf file (primary_maf)", "Cohort 2 maf file (relapse_maf)"] [maftools.paramters.readfiles.input.comparsion_input.choices] primary_maf = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(maf)', full.names = TRUE), featch_files(c('maf', 'maf.gz'))$file_path)}" relapse_maf = "!!glue {c(list.files(system.file('extdata', package = 'maftools'), '(maf)', full.names = TRUE), featch_files(c('maf', 'maf.gz'))$file_path)}" [maftools.paramters.readfiles.input.comparsion_input.selected] primary_maf = "@>@system.file('extdata', 'APL_primary.maf.gz', package = 'maftools')@<@" relapse_maf = "@>@system.file('extdata', 'APL_relapse.maf.gz', package = 'maftools')@<@" [maftools.paramters.readfiles.input.other_params] title = "Other paramters" title_control = "class = 'input-section-p'" varname = ["is_tcga"] input_id = ["maftools_is_tcga"] type = ["shiny::checkboxInput"] label = ["Reading file: Maf file is TCGA format? (is_tcga is TRUE)", "Command to read files."] [maftools.paramters.readfiles.input.other_params.value] is_tcga = true [maftools.paramters.readfiles.input.start_analysis] input_id = "start_maftools_analysis" type = "shiny::actionButton" label = "Run" [maftools.paramters.getFields] section_type = "output" rcmd_last = "getFields(laml)" render_type = "shiny::renderPrint" render_id = "maftools_fields_summary" output_type = "shiny::verbatimTextOutput" progressbar_message = "Maftools getFields" [maftools.paramters.getSampleSummary] section_type = "output" rcmd_last = "getSampleSummary(laml)" render_type = "DT::renderDataTable" render_id = "maftools_sample_summary" output_type = "DT::dataTableOutput" progressbar_message = "Maftools getSampleSummary" [maftools.paramters.getGeneSummary] section_type = "output" rcmd_last = "getGeneSummary(laml)" render_type = "DT::renderDataTable" render_id = "maftools_gene_summary" output_type = "DT::dataTableOutput" progressbar_message = "Maftools getGeneSummary" [maftools.paramters.getClinicalData] section_type = "output" render_type = "DT::renderDataTable" render_id = "maftools_clinical_data" output_type = "DT::dataTableOutput" rcmd_last = "getClinicalData(laml)" progressbar_message = "Maftools getClinicalData" [maftools.paramters.plotmafSummary] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_maf_summary" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 14, units='cm', bg='transparent'" rcmd_last = """plotmafSummary(maf = laml, rmOutlier = TRUE, addStat = 'median', dashboard = TRUE, titvRaw = FALSE)""" progressbar_message = "Maftools plotmafSummary" [maftools.paramters.oncoplot_default] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_oncoplots" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 14, units='cm', bg='transparent'" rcmd_last = "oncoplot(maf = laml, top = 10, fontSize = 12)" progressbar_message = "Maftools oncoplot_default" [maftools.paramters.oncoplot_with_cnv] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_oncoplots_cnv" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 14, units='cm', bg='transparent'" rcmd_last = "oncoplot(maf = laml.plus.gistic, top = 10, fontSize = 12)" progressbar_message = "Maftools oncoplot with CNV" [maftools.paramters.oncoplot_advanced] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_oncoplots_advanced" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 27, units='cm', bg='transparent'" rcmd_preprocess = """ col = RColorBrewer::brewer.pal(n = 8, name = 'Paired') names(col) = c('Frame_Shift_Del','Missense_Mutation', 'Nonsense_Mutation', 'Multi_Hit', 'Frame_Shift_Ins', 'In_Frame_Ins', 'Splice_Site', 'In_Frame_Del') #Color coding for FAB classification; try getAnnotations(x = laml) to see available annotations. fabcolors = RColorBrewer::brewer.pal(n = 8,name = 'Spectral') names(fabcolors) = c("M0", "M1", "M2", "M3", "M4", "M5", "M6", "M7") fabcolors = list(FAB_classification = fabcolors) mutsigQval = 0.01 clinicalFeatures = "FAB_classification" sortByAnnotation = TRUE oncoplot_advanced_params <- list(maf = laml, colors = col, mutsig = laml_mutsig, mutsigQval = mutsigQval, clinicalFeatures = clinicalFeatures, sortByAnnotation = sortByAnnotation, annotationColor = fabcolors) """ rcmd_last = """ do.call(oncoplot, oncoplot_advanced_params) """ progressbar_message = "Maftools oncoplot with advanced" [maftools.paramters.plotTiTv] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_titv" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'" rcmd_preprocess = "laml.titv = titv(maf = laml, plot = FALSE, useSyn = TRUE)" rcmd_last = "plotTiTv(res = laml.titv)" progressbar_message = "Maftools plotTiTv" [maftools.paramters.lollipopPlot] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_lollipop" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'" rcmd_last = """lollipopPlot(maf = laml, gene = 'DNMT3A', AACol = 'Protein_Change', showMutationRate = TRUE)""" progressbar_message = "Maftools lollipopPlot" [maftools.paramters.lollipopPlot2] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_lollipop2" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'" rcmd_last = """lollipopPlot2(m1 = primary_maf, m2 = relapse_maf, gene = "PML", AACol1 = "amino_acid_change", AACol2 = "amino_acid_change", m1_name = "Primary", m2_name = "Relapse")""" progressbar_message = "Maftools lollipopPlot2" [maftools.paramters.tcgaCompare] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_mutation_load" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'" rcmd_last = "laml.mutload = tcgaCompare(maf = laml, cohortName = 'Example-LAML')" progressbar_message = "Maftools tcgaCompare" [maftools.paramters.plotVaf] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_vaf_box" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'" rcmd_last = "plotVaf(maf = laml, vafCol = 'i_TumorVAF_WU')" progressbar_message = "Maftools plotVaf" [maftools.paramters.gisticChromPlot] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_gistic_genome" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'" rcmd_last = """gisticChromPlot(gistic = laml.gistic, markBands = "all")""" progressbar_message = "Maftools gisticChromPlot" [maftools.paramters.gisticBubblePlot] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_gistic_bubble" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'" rcmd_last = """gisticBubblePlot(gistic = laml.gistic)""" progressbar_message = "Maftools gisticBubblePlot" [maftools.paramters.gisticOncoPlot] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_gistic_oncoplot" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'" rcmd_last = """gisticOncoPlot(gistic = laml.gistic, clinicalData = getClinicalData(x = laml), clinicalFeatures = 'FAB_classification', sortByAnnotation = TRUE, top = 10)""" progressbar_message = "Maftools gisticOncoPlot" [maftools.paramters.somaticInteractions] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_somatic_inter" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'" rcmd_last = """somaticInteractions(maf = laml, top = 25, pvalue = c(0.05, 0.1))""" progressbar_message = "Maftools somaticInteractions" [maftools.paramters.oncostrip] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_somatic_inter_oncostrip" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'" rcmd_last = """oncostrip(maf = laml, genes = c('TP53', 'FLT3', 'RUNX1'))""" progressbar_message = "Maftools oncostrip" [maftools.paramters.plotOncodrive] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_oncodrive" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'" rcmd_preprocess = """laml.sig = oncodrive(maf = laml, AACol = 'Protein_Change', minMut = 5, pvalMethod = 'zscore')""" rcmd_last = """plotOncodrive(res = laml.sig, fdrCutOff = 0.1, useFraction = TRUE)""" progressbar_message = "Maftools plotOncodrive" [maftools.paramters.mafSurvival] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_survival" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'" rcmd_last = """mafSurvival(maf = laml, genes = 'DNMT3A', time = 'days_to_last_followup', Status = 'Overall_Survival_Status', isTCGA = TRUE)""" progressbar_message = "Maftools mafSurvival" [maftools.paramters.plotEnrichmentResults] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_clinical_enrichment" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'" rcmd_preprocess = "fab.ce = clinicalEnrichment(maf = laml, clinicalFeature = 'FAB_classification')" rcmd_last = """plotEnrichmentResults(enrich_res = fab.ce, pVal = 0.05)""" progressbar_message = "Maftools plotEnrichmentResults" [maftools.paramters.plotClusters] section_type = "output" render_type = "shiny::renderPlot" render_id = "maftools_plot_clusters" output_type = "shiny::plotOutput" export_engine = "Cairo" export_params = "type = 'pdf', width = 21, height = 17, units='cm', bg='transparent'" rcmd_preprocess = "tcga.ab.2972.het = inferHeterogeneity(maf = laml, tsb = 'TCGA-AB-2972', vafCol = 'i_TumorVAF_WU')" rcmd_last = "plotClusters(clusters = tcga.ab.2972.het)" progressbar_message = "Maftools maftools_plot_clusters"
More demos
In another project annovarR, we developed several plugins files that have not been included in BioInstaller package, such as gvmap, clusterProfiler, ANNOVAR, CEMiTool, vcfanno, annovarR, etc. It is noted that we do not want to limite users to write R codes for extending the R shiny functions. But using the plugin files to simultaneously generate UI and server code is a good choice to simplify the steps for adding new functions in a Shiny application.
Try the BioInstaller package in your browser
Any scripts or data that you put into this service are public.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.