In LieberInstitute/megadepth: megadepth: BigWig and BAM related utilities
knitr::opts_chunk$set(
collapse = TRUE,
comment = "#>",
fig.path = "man/figures/README-",
out.width = "100%"
)
megadepth












The goal of megadepth is to provide an R interface to the command line tool Megadepth for BigWig and BAM related utilities created by Christopher Wilks. This R package enables fast processing of BigWig files on downstream packages such as dasper and recount3. The Megadepth software also provides utilities for processing BAM files and extracting coverage information from them.
Here is an illustration on how fast megadepth is compared to other tools for processing local and remote BigWig files.
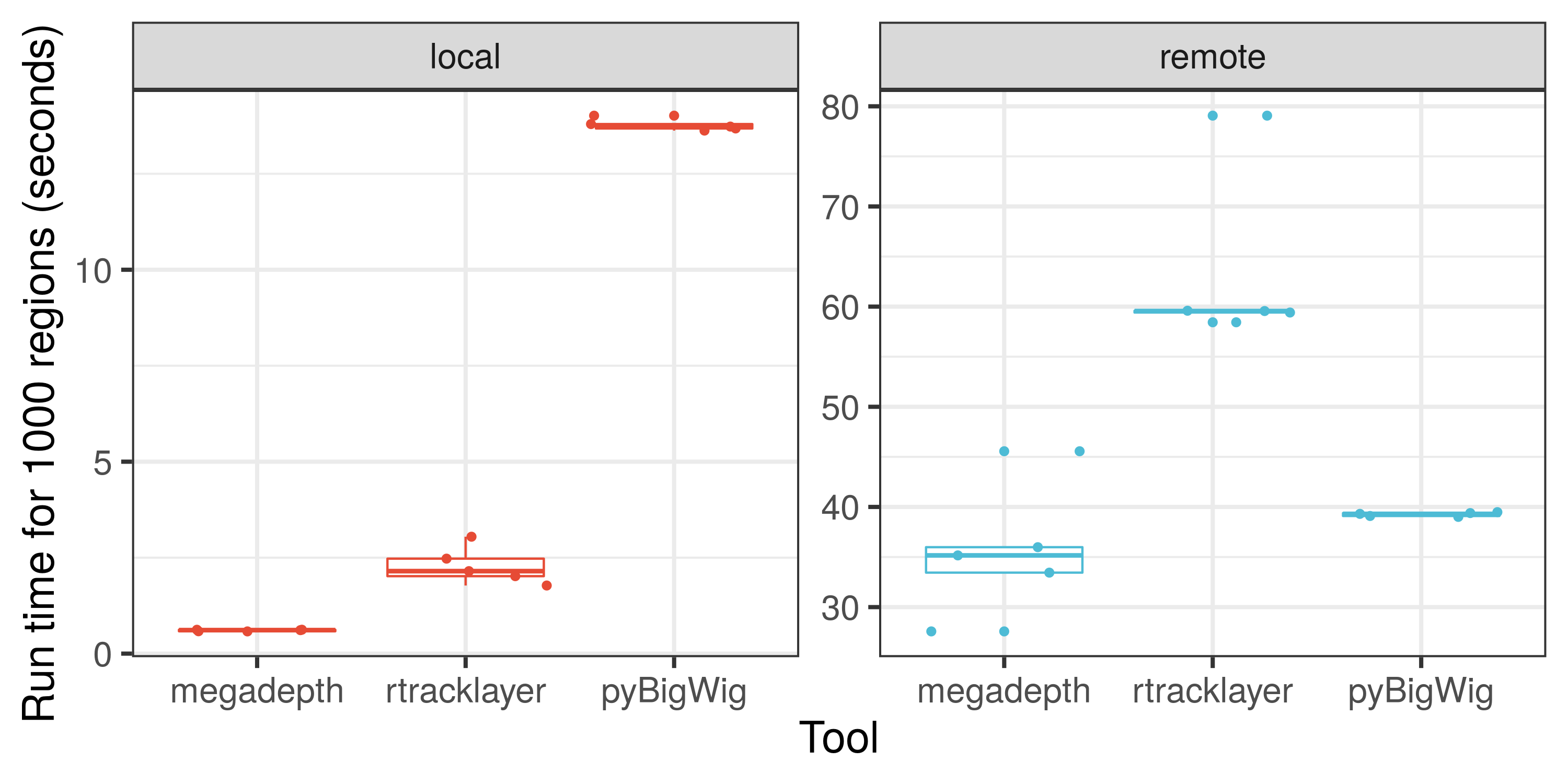
Throughout the documentation we use a capital M to refer to the software by Christopher Wilks and a lower case m to refer to this R/Bioconductor package.
Installation instructions
Get the latest stable R release from CRAN. Then install megadepth from Bioconductor using the following code:
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("megadepth")
And the development version from GitHub with:
BiocManager::install("LieberInstitute/megadepth")
Example
In the following example, we install Megadepth with install_megadepth(), which downloads a binary for your OS (Linux, Windows or macOS). We can then use with an example BigWig file to compute the coverage at a set of regions.
## Load the R package
library("megadepth")
## Install Megadepth's pre-compiled binary on your system
install_megadepth()
## Next, we locate the example BigWig and annotation files
example_bw <- system.file("tests", "test.bam.all.bw",
package = "megadepth", mustWork = TRUE
)
annotation_file <- system.file("tests", "testbw2.bed",
package = "megadepth", mustWork = TRUE
)
## We can then use megadepth to compute the coverage
bw_cov <- get_coverage(example_bw, op = "mean", annotation = annotation_file)
bw_cov
Full set of utilities
Megadepth is very powerful and can do a lot of different things. The R/Bioconductor package provides two functions for interfacing with Megadepth, megadepth_cmd() and megadepth_shell(). For the first one, megadepth_cmd(), you need to know the actual command syntax you want to use and format it accordingly. If you are more comfortable with R functions, megadepth_shell() uses r BiocStyle::CRANpkg("cmdfun") to power this interface and capture the standard output stream into R.
To make it easier to use, megadepth includes functions that simplify the number of arguments, read in the output files, and converts them into R/Bioconductor friendly objects, such as get_coverage() illustrated above.
We hope that you'll find megadepth and Megadepth useful for your work. If you are interested in checking how fast megadepth is, check out the speed analysis comparison against other tools. Note that the size of the files used and the number of genomic regions queried will affect the speed comparisons.
## R-like interface
## that captures the standard output into R
head(megadepth_shell(help = TRUE))
## Command-like interface
megadepth_cmd("--help")
x <- megadepth_shell(help = TRUE)
cat(paste0(x, "\n"))
Citation
Below is the citation output from using citation('megadepth') in R. Please
run this yourself to check for any updates on how to cite megadepth.
print(citation("megadepth"), bibtex = TRUE)
Please note that the megadepth was only made possible thanks to many other R and bioinformatics software authors, which are cited either in the vignettes and/or the paper(s) describing this package.
Code of Conduct
Please note that the megadepth project is released with a Contributor Code of Conduct. By contributing to this project, you agree to abide by its terms.
Development tools
- Continuous code testing is possible thanks to GitHub actions through
r BiocStyle::CRANpkg('usethis'), r BiocStyle::CRANpkg('remotes'), r BiocStyle::Githubpkg('r-hub/sysreqs') and r BiocStyle::CRANpkg('rcmdcheck') customized to use Bioconductor's docker containers and r BiocStyle::Biocpkg('BiocCheck').
- Code coverage assessment is possible thanks to codecov and
r BiocStyle::CRANpkg('covr').
- The documentation website is automatically updated thanks to
r BiocStyle::CRANpkg('pkgdown').
- The code is styled automatically thanks to
r BiocStyle::CRANpkg('styler').
- The documentation is formatted thanks to
r BiocStyle::CRANpkg('devtools') and r BiocStyle::CRANpkg('roxygen2').
For more details, check the dev directory.
This package was developed using r BiocStyle::Biocpkg('biocthis').
ReCount project
The main documentation website for all the recount3-related projects is available at recount.bio. Please check that website for more information about how this R/Bioconductor package and other tools are related to each other.
Teams involved
r BiocStyle::Biocpkg('megadepth') was made possible to David Zhang, the author of r BiocStyle::Biocpkg("dasper"), and a member of the Mina Ryten's lab at UCL.
The ReCount family involves the following teams:
- Ben Langmead's lab at JHU Computer Science
- Kasper Daniel Hansen's lab at JHBSPH Biostatistics Department
- Leonardo Collado-Torres and Andrew E. Jaffe from LIBD
- Abhinav Nellore's lab at OHSU
- Jeff Leek's lab at JHBSPH Biostatistics Deparment
- Data hosted by SciServer from IDIES at JHU
| | | | |
| --- | --- | --- | --- |
|  |
|  |
|  |
|  |
|
LieberInstitute/megadepth documentation built on Dec. 14, 2024, 3:30 p.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
knitr::opts_chunk$set( collapse = TRUE, comment = "#>", fig.path = "man/figures/README-", out.width = "100%" )
megadepth
![]()
![]()
![]()
The goal of megadepth is to provide an R interface to the command line tool Megadepth for BigWig and BAM related utilities created by Christopher Wilks. This R package enables fast processing of BigWig files on downstream packages such as dasper and recount3. The Megadepth software also provides utilities for processing BAM files and extracting coverage information from them.
Here is an illustration on how fast megadepth is compared to other tools for processing local and remote BigWig files.
Throughout the documentation we use a capital M to refer to the software by Christopher Wilks and a lower case m to refer to this R/Bioconductor package.
Installation instructions
Get the latest stable R release from CRAN. Then install megadepth from Bioconductor using the following code:
if (!requireNamespace("BiocManager", quietly = TRUE)) { install.packages("BiocManager") } BiocManager::install("megadepth")
And the development version from GitHub with:
BiocManager::install("LieberInstitute/megadepth")
Example
In the following example, we install Megadepth with install_megadepth(), which downloads a binary for your OS (Linux, Windows or macOS). We can then use with an example BigWig file to compute the coverage at a set of regions.
## Load the R package library("megadepth") ## Install Megadepth's pre-compiled binary on your system install_megadepth() ## Next, we locate the example BigWig and annotation files example_bw <- system.file("tests", "test.bam.all.bw", package = "megadepth", mustWork = TRUE ) annotation_file <- system.file("tests", "testbw2.bed", package = "megadepth", mustWork = TRUE ) ## We can then use megadepth to compute the coverage bw_cov <- get_coverage(example_bw, op = "mean", annotation = annotation_file) bw_cov
Full set of utilities
Megadepth is very powerful and can do a lot of different things. The R/Bioconductor package provides two functions for interfacing with Megadepth, megadepth_cmd() and megadepth_shell(). For the first one, megadepth_cmd(), you need to know the actual command syntax you want to use and format it accordingly. If you are more comfortable with R functions, megadepth_shell() uses r BiocStyle::CRANpkg("cmdfun") to power this interface and capture the standard output stream into R.
To make it easier to use, megadepth includes functions that simplify the number of arguments, read in the output files, and converts them into R/Bioconductor friendly objects, such as get_coverage() illustrated above.
We hope that you'll find megadepth and Megadepth useful for your work. If you are interested in checking how fast megadepth is, check out the speed analysis comparison against other tools. Note that the size of the files used and the number of genomic regions queried will affect the speed comparisons.
## R-like interface ## that captures the standard output into R head(megadepth_shell(help = TRUE)) ## Command-like interface megadepth_cmd("--help")
x <- megadepth_shell(help = TRUE) cat(paste0(x, "\n"))
Citation
Below is the citation output from using citation('megadepth') in R. Please
run this yourself to check for any updates on how to cite megadepth.
print(citation("megadepth"), bibtex = TRUE)
Please note that the megadepth was only made possible thanks to many other R and bioinformatics software authors, which are cited either in the vignettes and/or the paper(s) describing this package.
Code of Conduct
Please note that the megadepth project is released with a Contributor Code of Conduct. By contributing to this project, you agree to abide by its terms.
Development tools
- Continuous code testing is possible thanks to GitHub actions through
r BiocStyle::CRANpkg('usethis'),r BiocStyle::CRANpkg('remotes'),r BiocStyle::Githubpkg('r-hub/sysreqs')andr BiocStyle::CRANpkg('rcmdcheck')customized to use Bioconductor's docker containers andr BiocStyle::Biocpkg('BiocCheck'). - Code coverage assessment is possible thanks to codecov and
r BiocStyle::CRANpkg('covr'). - The documentation website is automatically updated thanks to
r BiocStyle::CRANpkg('pkgdown'). - The code is styled automatically thanks to
r BiocStyle::CRANpkg('styler'). - The documentation is formatted thanks to
r BiocStyle::CRANpkg('devtools')andr BiocStyle::CRANpkg('roxygen2').
For more details, check the dev directory.
This package was developed using r BiocStyle::Biocpkg('biocthis').
ReCount project
The main documentation website for all the recount3-related projects is available at recount.bio. Please check that website for more information about how this R/Bioconductor package and other tools are related to each other.
Teams involved
r BiocStyle::Biocpkg('megadepth') was made possible to David Zhang, the author of r BiocStyle::Biocpkg("dasper"), and a member of the Mina Ryten's lab at UCL.
The ReCount family involves the following teams:
- Ben Langmead's lab at JHU Computer Science
- Kasper Daniel Hansen's lab at JHBSPH Biostatistics Department
- Leonardo Collado-Torres and Andrew E. Jaffe from LIBD
- Abhinav Nellore's lab at OHSU
- Jeff Leek's lab at JHBSPH Biostatistics Deparment
- Data hosted by SciServer from IDIES at JHU
| | | | |
| --- | --- | --- | --- |
| | ![]() |
| ![]() |
| ![]() |
|
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.