README.md
In jpmeagher/vbar: Variational Bayes Ancestral Reconstruction
vbar
The goal of vbar is to implement Variational Bayes Ancestral
Reconstruction for collections of discrete and continuous phenotypic
traits given a phylogeny of evolutionary relationships between species.
Discrete traits may be ordinal or nominal, while continuous traits may
be scalar- or function-valued. Ancestral Reconstruction is based on the
Phylogenetic Latent Variable Model (PLVM) for trait evolution. This
model accommodates repeated measurements for extant species.
This readme is laid out as follows: after presenting code installing the
vbar package, the Generalised Phylogenetic Latent Variable Model
(PLVM) for trait evolution is outlined. In order to illustrate more
clearly the assumptions underpinning this model, a synthetic data set is
generated from the PLVM. Approximate Bayesian Inference for this
synthetic data set, which is included with the package, demonstrates how
the vbar package is applied for Ancestral Reconstruction.
For the code required to fit the generalised PLVM to data, skip to the
section on Approximate Bayesian
Inference
Installation
You can install the development version from
GitHub with:
# install.packages("devtools")
devtools::install_github("jpmeagher/vbar")
The Generalised Phylogenetic Latent Variable Model
Consider the
 -dimensional
observation of
-dimensional
observation of
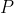 discrete- or continuous-valued manifest traits
discrete- or continuous-valued manifest traits
 ,
specific to individual
,
specific to individual
 within taxon
within taxon
 ,
where traits may be ordinal-, nominal-, scalar- or function-valued.
,
where traits may be ordinal-, nominal-, scalar- or function-valued.
The shared evolutionary history between taxa is modeled as a known,
fixed phylogeny
 .
.
 is a bifurcating, directed, acyclic graph tree with nodes
is a bifurcating, directed, acyclic graph tree with nodes
 linked by branches
linked by branches
 .
The graph originates at the degree-2 root node
.
The graph originates at the degree-2 root node
 and terminates at the degree-1 terminal nodes
and terminates at the degree-1 terminal nodes
 corresponding to the extant taxa. Node
corresponding to the extant taxa. Node
 is linked to its parent
is linked to its parent
 by the branch of length
by the branch of length
 for all
for all
 .
For notational convenience, we let
.
For notational convenience, we let
 denote a continuous position along a branch of
,
where
denote a continuous position along a branch of
,
where
 denotes the position of node
for all
denotes the position of node
for all
 .
Furthermore, the patristic distance operator
.
Furthermore, the patristic distance operator
 defines the shortest path from
defines the shortest path from
 to
to
 over
.
over
.
For each manifest trait, we introduce auxiliary traits
 such that
such that
 ,
there exists a deterministic map
,
there exists a deterministic map
 ,
and auxiliary traits mapping
,
and auxiliary traits mapping
 are indexed by
are indexed by
 .
This allows us to adopt a probit model for discrete traits.
.
This allows us to adopt a probit model for discrete traits.
If
 for
for
 is a nominal trait belonging to one of
is a nominal trait belonging to one of
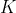 unordered categories, we have a
-to-1
mapping such that
unordered categories, we have a
-to-1
mapping such that

If
 for
is an ordinal trait belonging to one of
ordered categories, we have a 1-to-1 mapping
for
is an ordinal trait belonging to one of
ordered categories, we have a 1-to-1 mapping

given thresholds
 where
where
 ,
,
 ,
,
 ,
and
,
and
 .
Finally, if
takes continuous values, then any bijective map to the real numbers is
appropriate. We then assume that
.
Finally, if
takes continuous values, then any bijective map to the real numbers is
appropriate. We then assume that

where
 is the expected auxiliary trait and
is the expected auxiliary trait and
 is a diagonal precision matrix parameterised by
is a diagonal precision matrix parameterised by
 .
Note that we consider one independent precision parameter for each of
the
manifest traits, where
.
Note that we consider one independent precision parameter for each of
the
manifest traits, where
 when
when
 maps to the
maps to the
 manifest trait and
manifest trait and
 is fixed when that trait is discrete.
is fixed when that trait is discrete.
In order to define the PLVM, let

where
 is the loading matrix with columns denoted
is the loading matrix with columns denoted
 for
for
 ,
rows
,
rows
 for
for
 ,
and
,
and
 is the individual-specific latent trait. We link
is the individual-specific latent trait. We link
 to the taxon-specific latent trait
to the taxon-specific latent trait
 by setting
by setting

where
 is the standard deviation of the inter-taxon variation driven by
is the standard deviation of the inter-taxon variation driven by
 and we have
and we have
 .
Taxon-specific latent traits are modelled as independent phylogenetic
Ornstein-Uhlenbeck (OP) processes over
such that
.
Taxon-specific latent traits are modelled as independent phylogenetic
Ornstein-Uhlenbeck (OP) processes over
such that

where
 is the heritability of the
is the heritability of the
 latent trait such that
latent trait such that
 ,
,
 is the fixed phylogenetic length-scale, and
is the fixed phylogenetic length-scale, and

identifies extant taxa. Restricting
 and
and
 in this manner avoids scale invariance in the Gaussian likelihood, but
still provides a flexible model for latent trait evolution, similar to
the Phylogenetic Mixed Model for trait evolution.
in this manner avoids scale invariance in the Gaussian likelihood, but
still provides a flexible model for latent trait evolution, similar to
the Phylogenetic Mixed Model for trait evolution.
The OU process is a Gauss-Markov process. As such, it can be shown that

where, setting
 and
and
 ,
,

for
 .
We also define
.
We also define
 and
and
 at this point.
at this point.
The PLVM admits conjugate priors for the loading
 and free auxiliary trait precision parameters
and free auxiliary trait precision parameters
 when we have
when we have

where
 is the Automatic Relevance Determination (ARD) precision hyperparameter
such that
is the Automatic Relevance Determination (ARD) precision hyperparameter
such that
 ,
,
 is a fixed covariance matrix enforcing smoothness constraints on
is a fixed covariance matrix enforcing smoothness constraints on
 ,
and
,
and
 and
and
 are shape and rate hyperparameters for precision associated with trait
are shape and rate hyperparameters for precision associated with trait
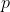 taking values along a continuum, and we let
taking values along a continuum, and we let
 denote the one-point distribution with all its mass at
denote the one-point distribution with all its mass at
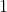 .
.
The PLVM also provides a direct approach to approach to ancestral
reconstruction. Firstly, note that this model for trait evolution allows
for within-taxon variation on the latent traits, governed by
 .
Stripping away this stochastic effect on trait evolution allows us
define taxon-specific manifest and auxiliary traits,
.
Stripping away this stochastic effect on trait evolution allows us
define taxon-specific manifest and auxiliary traits,
 and
and
 ,
such that
,
such that

for
,
 as before, and
as before, and
 is the diagonal covariance matrix parametrised by
is the diagonal covariance matrix parametrised by
 .
.
Sampling from the Generative Model
Before outlining our approach inference for the PLVM, we sample from the
model. This provides the synthetic data set we use to validate the
inference scheme. We require the following packages.
library(vbar)
library(ape)
library(ggplot2)
library(magrittr)
library(reshape2)
library(dplyr)
library(mvnfast)
library(HDInterval)
library(ggpubr)
Our analysis considers
 extant taxa with a shared evolutionary history defined by the phylogeny
below. We label the root node and its children for future reference.
Note that the
extant taxa with a shared evolutionary history defined by the phylogeny
below. We label the root node and its children for future reference.
Note that the ape package labels the root node
 by default.
by default.
S <- 2^7
set.seed(98)
phy <- rcoal(S) %>%
scale_phylo(max_dist = 1)

 traits are recorded for
traits are recorded for
 individuals in the extant taxon
.
These traits consist of an ordinal trait with
individuals in the extant taxon
.
These traits consist of an ordinal trait with
 ordered categories, a nominal trait with
ordered categories, a nominal trait with
 unordered categories, a scalar-valued continuous trait, and a strictly
positive function-valued trait registered to 32 equidistant points along
the unit interval. Thus,
unordered categories, a scalar-valued continuous trait, and a strictly
positive function-valued trait registered to 32 equidistant points along
the unit interval. Thus,
 and
and
 are
are
 and
and
 dimensional vectors respectively.
dimensional vectors respectively.
P <- 4
N_s <- rep(3, S)
N <- sum(N_s)
K_ord <- 4
K_nom <- 3
Auxiliary traits are generated by a PLVM with
 latent traits, parameterised by
and
latent traits, parameterised by
and
 such that auxiliary traits associated with discrete traits have
precision 1, the scalar-valued continuous trait has precision 10, and
the function-valued trait has precision of 10000. The loading matrix
is presented below.
such that auxiliary traits associated with discrete traits have
precision 1, the scalar-valued continuous trait has precision 10, and
the function-valued trait has precision of 10000. The loading matrix
is presented below.
auxiliary_trait_precision <- c(1, 1, 10, 10000)
L <- 3
# FVT Loading
D_fvt <- 2^5
x_fvt <- seq(0, 1, length.out = D_fvt)
loading_fvt_means <- c(.2, .5, .75)
loading_fvt_sds <- c(.06, .12, .07)
loading_fvt <- sapply(
seq_along(loading_fvt_means),
function(i) dnorm(x_fvt, mean = loading_fvt_means[i], sd = loading_fvt_sds[i])
)
loading_fvt <- loading_fvt %*% diag(1 / c(5, 4, 6))
# Continuous Trait Loading
loading_con <- c(0.5, 0.25, -0.25)
# Nominal Trait loading
loading_nom <- matrix(c(1, 0, 0, 0, -0.5, 0, 0, 0, 1), byrow = TRUE, nrow = K_nom, ncol = L)
# Ordinal Trait Loading
loading_ord <- c(0, 1, 0.5)
# Full Loading
loading <- rbind(loading_ord, loading_nom, loading_con, loading_fvt)
rownames(loading) <- c(
"ord",
paste("nom", 1:K_nom, sep = "."),
"con",
paste("fvt", 1:D_fvt, sep = ".")
)
colnames(loading) <- paste("loading", 1:L, sep = ".")

The distribution of taxon- and individual-specific latent traits is
defined by the phylogenetic hyperparameters
 and
and
 and so we sample
.
and so we sample
.
tau <- sqrt(c(0.001, 0.05, 0.1))
h <- sqrt(c(0.95, 0.66, 0.25))
set.seed(99)
taxon_specific_latent_traits <- sapply(
h, function(x) {
simulate_phylogenetic_ou(
phy = phy,
heritable_amplitude = x, length_scale = 2,
environmental_amplitude = sqrt(1 - x^2),
internal = TRUE
)
}
)
taxon_id <- sapply(1:S, function(i) rep(phy$tip.label[i], N_s[i])) %>%
c()
individual_specific_latent_traits <- sapply(
1:L, function(l){
sqrt(1 - tau[l]^2) * taxon_specific_latent_traits[taxon_id, l] +
tau[l] * rnorm(N)
})
Given
,
,
and
,
we can define
 and sample
.
and sample
.
D_prime <- nrow(loading)
auxiliary_traits_expectation <- individual_specific_latent_traits %*% t(loading)
auxiliary_trait_precision_vec <- c(
auxiliary_trait_precision[1],
rep(auxiliary_trait_precision[2], K_nom),
auxiliary_trait_precision[3],
rep(auxiliary_trait_precision[4], D_fvt)
)
set.seed(100)
auxiliary_traits <- auxiliary_traits_expectation +
t(
sapply(1:N, function(i){
rnorm(D_prime, sd = sqrt(1 / auxiliary_trait_precision_vec))
}
)
)
We map
to
by
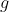 given the ordinal trait cut-off points
given the ordinal trait cut-off points
 .
Furthermore, we set
.
Furthermore, we set
 when the index
when the index
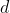 corresponds to a function-valued trait.
corresponds to a function-valued trait.
cut_off_points <- c(-Inf, 0, 0.5, 1, Inf)
auxiliary_index_ord <- 1
auxiliary_index_nom <- max(auxiliary_index_ord) + (1:K_nom)
auxiliary_index_con <- max(auxiliary_index_nom) + 1
auxiliary_index_fvt <- max(auxiliary_index_con) + 1:D_fvt
manifest_traits <- data.frame(
taxon_id = taxon_id,
ord = sapply(auxiliary_traits[, auxiliary_index_ord], function(x){
x > cut_off_points
}) %>%
colSums() %>%
factor(ordered = TRUE),
nom = apply(auxiliary_traits[, auxiliary_index_nom], 1, which.max) %>%
factor(ordered = FALSE),
con = auxiliary_traits[, auxiliary_index_con],
fvt = exp(unname(auxiliary_traits[, auxiliary_index_fvt]))
)

Given the mapping
,
loading
,
auxiliary trait precision parameters
,
individual-specific latent traits, and phylogenetic hyperparameters
and
 ,
we can explore the distribution of taxon-specific manifest traits
for any
.
,
we can explore the distribution of taxon-specific manifest traits
for any
.
covariance_ou <- ou_kernel(
dist.nodes(phy),
amplitude = 1, length_scale = 2
)
covariance_f <- lapply(h, function(x){
tmp <- x^2 * covariance_ou
tmp[1:S, 1:S] <- tmp[1:S, 1:S] + (1 - x^2)* diag(S)
colnames(tmp) <- rownames(tmp) <- c(phy$tip.label, S + (1:(S-1)))
tmp
})
covariance_zf <- lapply(1:L, function(i){
tmp <- (1 - tau[i]^2) * covariance_f[[i]]
tmp <- tmp[
c(taxon_id, phy$tip.label, S + (1:(S-1))),
c(taxon_id, phy$tip.label, S + (1:(S-1)))
]
tmp[1:N, 1:N] <- tmp[1:N, 1:N] + tau[i]^2 * diag(N)
tmp
})
ancestral_mean <- sapply(1:L, function(i){
(covariance_zf[[i]][-(1:N), (1:N)] %*%
chol2inv(chol(covariance_zf[[i]][(1:N), (1:N)])) %*%
individual_specific_latent_traits[, i])[, 1]
})
ancestral_cov <- lapply(1:L, function(i){
covariance_zf[[i]][-(1:N), -(1:N)] -
covariance_zf[[i]][-(1:N), (1:N)] %*%
chol2inv(chol(covariance_zf[[i]][(1:N), (1:N)])) %*%
covariance_zf[[i]][(1:N), -(1:N)]
})
ancestral_sd <- sapply(ancestral_cov, function(X){
sqrt(diag(X))
})
a <- "130" # ancestor
M <- 1000 # samples for ancestral reconstruction
scaled_taxon_latent_trait_ar <- rmvn(
M, mu = ancestral_mean[a,], sigma = diag(ancestral_sd[a, ]), isChol = TRUE
)
auxiliary_trait_ar <- scaled_taxon_latent_trait_ar %*% t(loading) +
t(
sapply(1:M, function(i){
rnorm(D_prime, sd = sqrt(1 / auxiliary_trait_precision_vec))
}
)
)
manifest_trait_ar <- data.frame(
ord = sapply(auxiliary_trait_ar[, auxiliary_index_ord], function(x){
x > cut_off_points
}) %>%
colSums() %>%
factor(ordered = TRUE),
nom = apply(auxiliary_trait_ar[, auxiliary_index_nom], 1, which.max) %>%
factor(ordered = FALSE),
con = auxiliary_trait_ar[, auxiliary_index_con],
fvt = exp(unname(auxiliary_trait_ar[, auxiliary_index_fvt]))
)

Approximate Bayesian Inference
Our objective is to infer a posterior distribution over
individual-specific auxiliary traits, the PLVM loading, individual- and
taxon-specific latent traits, and free auxiliary trait precision
parameters given manifest traits, a fixed phylogeny, and priors on the
model parameters. That is

which can be expressed as

where we have introduced the notation
 for
for
 which suppresses the index
which suppresses the index
 ,
,
 and
and
 have analogous definitions, and
have analogous definitions, and
 model hyperparameters.
and
model hyperparameters.
and
 are used interchangeably as is most convenient. This hierarchical model
is defined by the distributions over manifest and auxiliary variables
are used interchangeably as is most convenient. This hierarchical model
is defined by the distributions over manifest and auxiliary variables

latent variables

and parameters

where
is the diagonal covariance matrix parametrised by
,
 is the diagonal covariance matrix parametrised by
.
is the diagonal covariance matrix parametrised by
.
A Co-ordinate Ascent Variational Inference (CAVI) algorithm allows
approximate Bayesian Inference for this model. To implement this
inference scheme we use the vbar package.
We start by decluttering the global environment.
rm(list = ls())
The synthetic data set of manifest traits for
 individuals across
extant taxa is included with the
individuals across
extant taxa is included with the vbar package. It consists of 5
variables. That is an id variable taxon_id alongside
traits, namely the ordinal trait ord, the nominal trait nom, the
continuous trait con, and the function-valued trait fvt. Note that
the fvt variable is a list of lists, such that each element consists
of a numeric vector of length 32.
synthetic_traits %>% str(list.len = 5)
#> 'data.frame': 384 obs. of 5 variables:
#> $ taxon_id: chr "t18" "t18" "t18" "t104" ...
#> $ ord : Ord.factor w/ 4 levels "1"<"2"<"3"<"4": 1 3 1 1 2 2 1 1 2 4 ...
#> $ nom : Factor w/ 3 levels "1","2","3": 3 1 3 3 3 2 2 2 1 3 ...
#> $ con : num -0.0441 0.3153 0.2793 -0.3384 -0.3122 ...
#> $ fvt :List of 384
#> ..$ : num 1.002 0.989 0.988 0.937 0.881 ...
#> ..$ : num 0.981 1 0.972 0.947 0.86 ...
#> ..$ : num 0.979 1.004 0.981 0.933 0.868 ...
#> ..$ : num 1.007 0.982 0.938 0.85 0.671 ...
#> ..$ : num 0.997 0.987 0.938 0.837 0.679 ...
#> .. [list output truncated]
Our analysis requires a data frame where each row consists of an id
variable alongside a
-dimensional
vector of traits.
manifest_trait_df <- cbind(synthetic_traits[, 1:4], fvt = t(simplify2array(synthetic_traits$fvt)))
The first step required for CAVI is to specify metadata for the manifest
traits using the specify_manifest_trait_metadata function. This
function allows the user to specify the specific properties of a
particular data set and, given the data frame containing manifest
traits, performs a number of tests to verify that the metadata matches
the manifest traits.
# ?specify_manifest_trait_metadata
metadata <- specify_manifest_trait_metadata(
n_traits = 4,
trait_names = colnames(synthetic_traits)[-1],
trait_type = factor(c("ord", "nom", "con", "fvt")),
trait_levels = c(
nlevels(manifest_trait_df$ord), nlevels(manifest_trait_df$nom),
NA, NA
),
manifest_trait_index = list(
ord = 2, nom = 3, con = 4, fvt = 4 + 1:32
),
auxiliary_trait_index = list(
ord = 1, nom = 1 + 1:3, con = 5, fvt = 5 + 1:32
),
link_functions = list(
ord = ordinal_link, nom = nominal_link, con = function(x) x, fvt = function(x) exp(x)
),
inverse_link_functions = list(
ord = function(y){
ordinal_inverse_link(
y, cut_off_points = metadata$cut_off_points$ord,
mu = rep(0, nrow(manifest_trait_df)), return_expectation = FALSE
)
},
nom = function(y){
nominal_inverse_link(
y, mu = matrix(0, nrow(manifest_trait_df), length(metadata$categories$nom)),
n_samples = 1000, return_expectation = FALSE
)
},
con = function(y) y,
fvt = function(y) log(data.matrix(y))
),
cut_off_points = list(
ord = c(-Inf, 0, sort(runif(nlevels(manifest_trait_df$ord) - 2)), Inf),
nom = NA, con = NA, fvt = NA
),
categories = list(
ord = NA, nom = levels(manifest_trait_df$nom),
con = NA, fvt = NA
),
manifest_trait_df = manifest_trait_df,
perform_checks = TRUE
)
Note that the exponential link function for fvt maps real valued
auxiliary traits to positive real valued manifest traits. In addition,
the link functions specified here are only required to initialise the
PLVM. CAVI will allow the
Given the metadata, we are now ready to initialise a PLVM using
initialise_plvm. This requires the phylogeny of evolutionary
relationships between extant taxa, the dimension of the latent trait,
and a prior correlation structure for the columns of the PLVM Loading.
# ?initialise_plvm
L <- 3
C_w <- metadata$auxiliary_trait_index %>%
unlist() %>%
length() %>%
diag()
x_fvt <- seq(0, 1, length.out = length(metadata$auxiliary_trait_index$fvt))
d <- abs(outer(x_fvt, x_fvt, "-"))
ell <- 1 / (2 * pi) # Length-scale to be specified by the user
C_w[metadata$auxiliary_trait_index$fvt, metadata$auxiliary_trait_index$fvt] <- (exp_quad_kernel(d, 1, ell) + (1e-6 * diag(length(x_fvt)))) / (1 + 1e-6)
plvm <- initialise_plvm(
manifest_trait_df = manifest_trait_df,
metadata = metadata,
L = L,
phy = synthetic_trait_model_specification$phylogeny,
id_label = "taxon_id",
loading_prior_correlation = C_w)
This is the minimum required specification for vbar to initialise a
PLVM. By default, auxiliary traits are sampled at random given the
initial ordinal trait cut-off points and discrete manifest traits.
Varimax analysis of the resulting auxiliary traits provide sensible
initial values for the PLVM Loading. All the remaining parameters and
latent variables are then sampled at random.
Given an initial PLVM, we can implement CAVI using the cavi_plvm
function. In the interest of brevity, the CAVI algorithm is run for 10
iterations only. For empirical data problems we would iterate to
convergence of the ELBO, defined by tol.
cavi <- cavi_plvm(
plvm_list = plvm,
tol = 1e-1, max_iter = 10,
n_samples = 1000, random_seed = 202,
progress_bar = TRUE,
perform_checks = FALSE
)
#> | | | 0% | |======= | 10% | |============== | 20% | |===================== | 30% | |============================ | 40% | |=================================== | 50% | |========================================== | 60% | |================================================= | 70% | |======================================================== | 80% | |=============================================================== | 90% | |======================================================================| 100%
We can compare the output from the CAVI for the PLVM with the known
parameters of the generative model.
First, we verify that the CAVI algorithm is maximising the ELBO. The
figure omits the ELBO at the first 5 iterations of the CAVI for clarity.

We then account of any reordering or reflection of the inferred loadings
relative to the true model specification.
R_mat <- IMIFA::Procrustes(
cavi$model$loading_expectation,
synthetic_trait_model_specification$loading
)$R %>% round
We can then compare inferred loadings (error bars and dotted lines) to
the model specification (points and solid lines). This allows us to
verify that

These are broadly similar, although the uncertainty intervals do not
necessarily cover the model specification. There are a number of reasons
for this. Variational Inference systematically underestimate
uncertainty, and it can be difficult to infer loadings for discrete
traits. The most relevant issue however, is that
and
are non-identifiable in this model and so the output from CAVI is
sensitive to initial values.
The primary goal of fitting this model to data is to perform Ancestral
Reconstruction and so we will compare the approximate distribution for
ancestral traits to those specified by the model. We examine the
ancestral reconstruction of the ancestor at node 130 of the phylogeny.
This is the same node for which the true ancestral distributions are
presented above.
a <- 130
approx_ar <- variational_ancestral_reconstruction(
taxon_specific_latent_trait_expectation = cavi$model$taxon_specific_latent_trait_expectation[a, ],
taxon_specific_latent_trait_covariance = cavi$model$taxon_specific_latent_trait_covariance[a, ],
loading_expectation = cavi$model$loading_expectation,
precision_vector = cavi$model$precision_vector,
metadata = cavi$model$metadata
)

Comparing this approximation to the true ancestral distribution, we find
that they are broadly similar, although the approximate Bayesian
inference scheme will tend to underestimate the uncertainty associated
with ancestral traits.
Conclusion
The Co-ordinate Ascent Variational Inference scheme for approximate
Bayesian inference on a generalised Phylogenetic Latent Variable Model
provides estimates for the ancestral distribution of any collection of
discrete and continuous traits.
jpmeagher/vbar documentation built on Nov. 22, 2022, 5:48 a.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
vbar
The goal of vbar is to implement Variational Bayes Ancestral
Reconstruction for collections of discrete and continuous phenotypic
traits given a phylogeny of evolutionary relationships between species.
Discrete traits may be ordinal or nominal, while continuous traits may
be scalar- or function-valued. Ancestral Reconstruction is based on the
Phylogenetic Latent Variable Model (PLVM) for trait evolution. This
model accommodates repeated measurements for extant species.
This readme is laid out as follows: after presenting code installing the
vbar package, the Generalised Phylogenetic Latent Variable Model
(PLVM) for trait evolution is outlined. In order to illustrate more
clearly the assumptions underpinning this model, a synthetic data set is
generated from the PLVM. Approximate Bayesian Inference for this
synthetic data set, which is included with the package, demonstrates how
the vbar package is applied for Ancestral Reconstruction.
For the code required to fit the generalised PLVM to data, skip to the section on Approximate Bayesian Inference
Installation
You can install the development version from GitHub with:
# install.packages("devtools")
devtools::install_github("jpmeagher/vbar")
The Generalised Phylogenetic Latent Variable Model
Consider the
-dimensional
observation of
discrete- or continuous-valued manifest traits
,
specific to individual
within taxon
,
where traits may be ordinal-, nominal-, scalar- or function-valued.
The shared evolutionary history between taxa is modeled as a known,
fixed phylogeny
.
is a bifurcating, directed, acyclic graph tree with nodes
linked by branches
.
The graph originates at the degree-2 root node
and terminates at the degree-1 terminal nodes
corresponding to the extant taxa. Node
is linked to its parent
by the branch of length
for all
.
For notational convenience, we let
denote a continuous position along a branch of
,
where
denotes the position of node
for all
.
Furthermore, the patristic distance operator
defines the shortest path from
to
over
.
For each manifest trait, we introduce auxiliary traits
such that
,
there exists a deterministic map
,
and auxiliary traits mapping
are indexed by
.
This allows us to adopt a probit model for discrete traits.
If
for
is a nominal trait belonging to one of
unordered categories, we have a
-to-1
mapping such that
If
for
is an ordinal trait belonging to one of
ordered categories, we have a 1-to-1 mapping
given thresholds
where
,
,
,
and
.
Finally, if
takes continuous values, then any bijective map to the real numbers is
appropriate. We then assume that
where
is the expected auxiliary trait and
is a diagonal precision matrix parameterised by
.
Note that we consider one independent precision parameter for each of
the
manifest traits, where
when
maps to the
manifest trait and
is fixed when that trait is discrete.
In order to define the PLVM, let
where
is the loading matrix with columns denoted
for
,
rows
for
,
and
is the individual-specific latent trait. We link
to the taxon-specific latent trait
by setting
where
is the standard deviation of the inter-taxon variation driven by
and we have
.
Taxon-specific latent traits are modelled as independent phylogenetic
Ornstein-Uhlenbeck (OP) processes over
such that
where
is the heritability of the
latent trait such that
,
is the fixed phylogenetic length-scale, and
identifies extant taxa. Restricting
and
in this manner avoids scale invariance in the Gaussian likelihood, but
still provides a flexible model for latent trait evolution, similar to
the Phylogenetic Mixed Model for trait evolution.
The OU process is a Gauss-Markov process. As such, it can be shown that
where, setting
and
,
for
.
We also define
and
at this point.
The PLVM admits conjugate priors for the loading
and free auxiliary trait precision parameters
when we have
where
is the Automatic Relevance Determination (ARD) precision hyperparameter
such that
,
is a fixed covariance matrix enforcing smoothness constraints on
,
and
and
are shape and rate hyperparameters for precision associated with trait
taking values along a continuum, and we let
denote the one-point distribution with all its mass at
.
The PLVM also provides a direct approach to approach to ancestral
reconstruction. Firstly, note that this model for trait evolution allows
for within-taxon variation on the latent traits, governed by
.
Stripping away this stochastic effect on trait evolution allows us
define taxon-specific manifest and auxiliary traits,
and
,
such that
for
,
as before, and
is the diagonal covariance matrix parametrised by
.
Sampling from the Generative Model
Before outlining our approach inference for the PLVM, we sample from the model. This provides the synthetic data set we use to validate the inference scheme. We require the following packages.
library(vbar)
library(ape)
library(ggplot2)
library(magrittr)
library(reshape2)
library(dplyr)
library(mvnfast)
library(HDInterval)
library(ggpubr)
Our analysis considers
extant taxa with a shared evolutionary history defined by the phylogeny
below. We label the root node and its children for future reference.
Note that the
ape package labels the root node
by default.
S <- 2^7
set.seed(98)
phy <- rcoal(S) %>%
scale_phylo(max_dist = 1)
traits are recorded for
individuals in the extant taxon
.
These traits consist of an ordinal trait with
ordered categories, a nominal trait with
unordered categories, a scalar-valued continuous trait, and a strictly
positive function-valued trait registered to 32 equidistant points along
the unit interval. Thus,
and
are
and
dimensional vectors respectively.
P <- 4
N_s <- rep(3, S)
N <- sum(N_s)
K_ord <- 4
K_nom <- 3
Auxiliary traits are generated by a PLVM with
latent traits, parameterised by
and
such that auxiliary traits associated with discrete traits have
precision 1, the scalar-valued continuous trait has precision 10, and
the function-valued trait has precision of 10000. The loading matrix
is presented below.
auxiliary_trait_precision <- c(1, 1, 10, 10000)
L <- 3
# FVT Loading
D_fvt <- 2^5
x_fvt <- seq(0, 1, length.out = D_fvt)
loading_fvt_means <- c(.2, .5, .75)
loading_fvt_sds <- c(.06, .12, .07)
loading_fvt <- sapply(
seq_along(loading_fvt_means),
function(i) dnorm(x_fvt, mean = loading_fvt_means[i], sd = loading_fvt_sds[i])
)
loading_fvt <- loading_fvt %*% diag(1 / c(5, 4, 6))
# Continuous Trait Loading
loading_con <- c(0.5, 0.25, -0.25)
# Nominal Trait loading
loading_nom <- matrix(c(1, 0, 0, 0, -0.5, 0, 0, 0, 1), byrow = TRUE, nrow = K_nom, ncol = L)
# Ordinal Trait Loading
loading_ord <- c(0, 1, 0.5)
# Full Loading
loading <- rbind(loading_ord, loading_nom, loading_con, loading_fvt)
rownames(loading) <- c(
"ord",
paste("nom", 1:K_nom, sep = "."),
"con",
paste("fvt", 1:D_fvt, sep = ".")
)
colnames(loading) <- paste("loading", 1:L, sep = ".")
The distribution of taxon- and individual-specific latent traits is
defined by the phylogenetic hyperparameters
and
and so we sample
.
tau <- sqrt(c(0.001, 0.05, 0.1))
h <- sqrt(c(0.95, 0.66, 0.25))
set.seed(99)
taxon_specific_latent_traits <- sapply(
h, function(x) {
simulate_phylogenetic_ou(
phy = phy,
heritable_amplitude = x, length_scale = 2,
environmental_amplitude = sqrt(1 - x^2),
internal = TRUE
)
}
)
taxon_id <- sapply(1:S, function(i) rep(phy$tip.label[i], N_s[i])) %>%
c()
individual_specific_latent_traits <- sapply(
1:L, function(l){
sqrt(1 - tau[l]^2) * taxon_specific_latent_traits[taxon_id, l] +
tau[l] * rnorm(N)
})
Given
,
,
and
,
we can define
and sample
.
D_prime <- nrow(loading)
auxiliary_traits_expectation <- individual_specific_latent_traits %*% t(loading)
auxiliary_trait_precision_vec <- c(
auxiliary_trait_precision[1],
rep(auxiliary_trait_precision[2], K_nom),
auxiliary_trait_precision[3],
rep(auxiliary_trait_precision[4], D_fvt)
)
set.seed(100)
auxiliary_traits <- auxiliary_traits_expectation +
t(
sapply(1:N, function(i){
rnorm(D_prime, sd = sqrt(1 / auxiliary_trait_precision_vec))
}
)
)
We map
to
by
given the ordinal trait cut-off points
.
Furthermore, we set
when the index
corresponds to a function-valued trait.
cut_off_points <- c(-Inf, 0, 0.5, 1, Inf)
auxiliary_index_ord <- 1
auxiliary_index_nom <- max(auxiliary_index_ord) + (1:K_nom)
auxiliary_index_con <- max(auxiliary_index_nom) + 1
auxiliary_index_fvt <- max(auxiliary_index_con) + 1:D_fvt
manifest_traits <- data.frame(
taxon_id = taxon_id,
ord = sapply(auxiliary_traits[, auxiliary_index_ord], function(x){
x > cut_off_points
}) %>%
colSums() %>%
factor(ordered = TRUE),
nom = apply(auxiliary_traits[, auxiliary_index_nom], 1, which.max) %>%
factor(ordered = FALSE),
con = auxiliary_traits[, auxiliary_index_con],
fvt = exp(unname(auxiliary_traits[, auxiliary_index_fvt]))
)
Given the mapping
,
loading
,
auxiliary trait precision parameters
,
individual-specific latent traits, and phylogenetic hyperparameters
and
,
we can explore the distribution of taxon-specific manifest traits
for any
.
covariance_ou <- ou_kernel(
dist.nodes(phy),
amplitude = 1, length_scale = 2
)
covariance_f <- lapply(h, function(x){
tmp <- x^2 * covariance_ou
tmp[1:S, 1:S] <- tmp[1:S, 1:S] + (1 - x^2)* diag(S)
colnames(tmp) <- rownames(tmp) <- c(phy$tip.label, S + (1:(S-1)))
tmp
})
covariance_zf <- lapply(1:L, function(i){
tmp <- (1 - tau[i]^2) * covariance_f[[i]]
tmp <- tmp[
c(taxon_id, phy$tip.label, S + (1:(S-1))),
c(taxon_id, phy$tip.label, S + (1:(S-1)))
]
tmp[1:N, 1:N] <- tmp[1:N, 1:N] + tau[i]^2 * diag(N)
tmp
})
ancestral_mean <- sapply(1:L, function(i){
(covariance_zf[[i]][-(1:N), (1:N)] %*%
chol2inv(chol(covariance_zf[[i]][(1:N), (1:N)])) %*%
individual_specific_latent_traits[, i])[, 1]
})
ancestral_cov <- lapply(1:L, function(i){
covariance_zf[[i]][-(1:N), -(1:N)] -
covariance_zf[[i]][-(1:N), (1:N)] %*%
chol2inv(chol(covariance_zf[[i]][(1:N), (1:N)])) %*%
covariance_zf[[i]][(1:N), -(1:N)]
})
ancestral_sd <- sapply(ancestral_cov, function(X){
sqrt(diag(X))
})
a <- "130" # ancestor
M <- 1000 # samples for ancestral reconstruction
scaled_taxon_latent_trait_ar <- rmvn(
M, mu = ancestral_mean[a,], sigma = diag(ancestral_sd[a, ]), isChol = TRUE
)
auxiliary_trait_ar <- scaled_taxon_latent_trait_ar %*% t(loading) +
t(
sapply(1:M, function(i){
rnorm(D_prime, sd = sqrt(1 / auxiliary_trait_precision_vec))
}
)
)
manifest_trait_ar <- data.frame(
ord = sapply(auxiliary_trait_ar[, auxiliary_index_ord], function(x){
x > cut_off_points
}) %>%
colSums() %>%
factor(ordered = TRUE),
nom = apply(auxiliary_trait_ar[, auxiliary_index_nom], 1, which.max) %>%
factor(ordered = FALSE),
con = auxiliary_trait_ar[, auxiliary_index_con],
fvt = exp(unname(auxiliary_trait_ar[, auxiliary_index_fvt]))
)
Approximate Bayesian Inference
Our objective is to infer a posterior distribution over individual-specific auxiliary traits, the PLVM loading, individual- and taxon-specific latent traits, and free auxiliary trait precision parameters given manifest traits, a fixed phylogeny, and priors on the model parameters. That is
which can be expressed as
where we have introduced the notation
for
which suppresses the index
,
and
have analogous definitions, and
model hyperparameters.
and
are used interchangeably as is most convenient. This hierarchical model
is defined by the distributions over manifest and auxiliary variables
latent variables
and parameters
where
is the diagonal covariance matrix parametrised by
,
is the diagonal covariance matrix parametrised by
.
A Co-ordinate Ascent Variational Inference (CAVI) algorithm allows
approximate Bayesian Inference for this model. To implement this
inference scheme we use the vbar package.
We start by decluttering the global environment.
rm(list = ls())
The synthetic data set of manifest traits for
individuals across
extant taxa is included with the
vbar package. It consists of 5
variables. That is an id variable taxon_id alongside
traits, namely the ordinal trait
ord, the nominal trait nom, the
continuous trait con, and the function-valued trait fvt. Note that
the fvt variable is a list of lists, such that each element consists
of a numeric vector of length 32.
synthetic_traits %>% str(list.len = 5)
#> 'data.frame': 384 obs. of 5 variables:
#> $ taxon_id: chr "t18" "t18" "t18" "t104" ...
#> $ ord : Ord.factor w/ 4 levels "1"<"2"<"3"<"4": 1 3 1 1 2 2 1 1 2 4 ...
#> $ nom : Factor w/ 3 levels "1","2","3": 3 1 3 3 3 2 2 2 1 3 ...
#> $ con : num -0.0441 0.3153 0.2793 -0.3384 -0.3122 ...
#> $ fvt :List of 384
#> ..$ : num 1.002 0.989 0.988 0.937 0.881 ...
#> ..$ : num 0.981 1 0.972 0.947 0.86 ...
#> ..$ : num 0.979 1.004 0.981 0.933 0.868 ...
#> ..$ : num 1.007 0.982 0.938 0.85 0.671 ...
#> ..$ : num 0.997 0.987 0.938 0.837 0.679 ...
#> .. [list output truncated]
Our analysis requires a data frame where each row consists of an id
variable alongside a
-dimensional
vector of traits.
manifest_trait_df <- cbind(synthetic_traits[, 1:4], fvt = t(simplify2array(synthetic_traits$fvt)))
The first step required for CAVI is to specify metadata for the manifest
traits using the specify_manifest_trait_metadata function. This
function allows the user to specify the specific properties of a
particular data set and, given the data frame containing manifest
traits, performs a number of tests to verify that the metadata matches
the manifest traits.
# ?specify_manifest_trait_metadata
metadata <- specify_manifest_trait_metadata(
n_traits = 4,
trait_names = colnames(synthetic_traits)[-1],
trait_type = factor(c("ord", "nom", "con", "fvt")),
trait_levels = c(
nlevels(manifest_trait_df$ord), nlevels(manifest_trait_df$nom),
NA, NA
),
manifest_trait_index = list(
ord = 2, nom = 3, con = 4, fvt = 4 + 1:32
),
auxiliary_trait_index = list(
ord = 1, nom = 1 + 1:3, con = 5, fvt = 5 + 1:32
),
link_functions = list(
ord = ordinal_link, nom = nominal_link, con = function(x) x, fvt = function(x) exp(x)
),
inverse_link_functions = list(
ord = function(y){
ordinal_inverse_link(
y, cut_off_points = metadata$cut_off_points$ord,
mu = rep(0, nrow(manifest_trait_df)), return_expectation = FALSE
)
},
nom = function(y){
nominal_inverse_link(
y, mu = matrix(0, nrow(manifest_trait_df), length(metadata$categories$nom)),
n_samples = 1000, return_expectation = FALSE
)
},
con = function(y) y,
fvt = function(y) log(data.matrix(y))
),
cut_off_points = list(
ord = c(-Inf, 0, sort(runif(nlevels(manifest_trait_df$ord) - 2)), Inf),
nom = NA, con = NA, fvt = NA
),
categories = list(
ord = NA, nom = levels(manifest_trait_df$nom),
con = NA, fvt = NA
),
manifest_trait_df = manifest_trait_df,
perform_checks = TRUE
)
Note that the exponential link function for fvt maps real valued
auxiliary traits to positive real valued manifest traits. In addition,
the link functions specified here are only required to initialise the
PLVM. CAVI will allow the
Given the metadata, we are now ready to initialise a PLVM using
initialise_plvm. This requires the phylogeny of evolutionary
relationships between extant taxa, the dimension of the latent trait,
and a prior correlation structure for the columns of the PLVM Loading.
# ?initialise_plvm
L <- 3
C_w <- metadata$auxiliary_trait_index %>%
unlist() %>%
length() %>%
diag()
x_fvt <- seq(0, 1, length.out = length(metadata$auxiliary_trait_index$fvt))
d <- abs(outer(x_fvt, x_fvt, "-"))
ell <- 1 / (2 * pi) # Length-scale to be specified by the user
C_w[metadata$auxiliary_trait_index$fvt, metadata$auxiliary_trait_index$fvt] <- (exp_quad_kernel(d, 1, ell) + (1e-6 * diag(length(x_fvt)))) / (1 + 1e-6)
plvm <- initialise_plvm(
manifest_trait_df = manifest_trait_df,
metadata = metadata,
L = L,
phy = synthetic_trait_model_specification$phylogeny,
id_label = "taxon_id",
loading_prior_correlation = C_w)
This is the minimum required specification for vbar to initialise a
PLVM. By default, auxiliary traits are sampled at random given the
initial ordinal trait cut-off points and discrete manifest traits.
Varimax analysis of the resulting auxiliary traits provide sensible
initial values for the PLVM Loading. All the remaining parameters and
latent variables are then sampled at random.
Given an initial PLVM, we can implement CAVI using the cavi_plvm
function. In the interest of brevity, the CAVI algorithm is run for 10
iterations only. For empirical data problems we would iterate to
convergence of the ELBO, defined by tol.
cavi <- cavi_plvm(
plvm_list = plvm,
tol = 1e-1, max_iter = 10,
n_samples = 1000, random_seed = 202,
progress_bar = TRUE,
perform_checks = FALSE
)
#> | | | 0% | |======= | 10% | |============== | 20% | |===================== | 30% | |============================ | 40% | |=================================== | 50% | |========================================== | 60% | |================================================= | 70% | |======================================================== | 80% | |=============================================================== | 90% | |======================================================================| 100%
We can compare the output from the CAVI for the PLVM with the known parameters of the generative model.
First, we verify that the CAVI algorithm is maximising the ELBO. The figure omits the ELBO at the first 5 iterations of the CAVI for clarity.
We then account of any reordering or reflection of the inferred loadings relative to the true model specification.
R_mat <- IMIFA::Procrustes(
cavi$model$loading_expectation,
synthetic_trait_model_specification$loading
)$R %>% round
We can then compare inferred loadings (error bars and dotted lines) to the model specification (points and solid lines). This allows us to verify that
These are broadly similar, although the uncertainty intervals do not
necessarily cover the model specification. There are a number of reasons
for this. Variational Inference systematically underestimate
uncertainty, and it can be difficult to infer loadings for discrete
traits. The most relevant issue however, is that
and
are non-identifiable in this model and so the output from CAVI is
sensitive to initial values.
The primary goal of fitting this model to data is to perform Ancestral Reconstruction and so we will compare the approximate distribution for ancestral traits to those specified by the model. We examine the ancestral reconstruction of the ancestor at node 130 of the phylogeny. This is the same node for which the true ancestral distributions are presented above.
a <- 130
approx_ar <- variational_ancestral_reconstruction(
taxon_specific_latent_trait_expectation = cavi$model$taxon_specific_latent_trait_expectation[a, ],
taxon_specific_latent_trait_covariance = cavi$model$taxon_specific_latent_trait_covariance[a, ],
loading_expectation = cavi$model$loading_expectation,
precision_vector = cavi$model$precision_vector,
metadata = cavi$model$metadata
)
Comparing this approximation to the true ancestral distribution, we find that they are broadly similar, although the approximate Bayesian inference scheme will tend to underestimate the uncertainty associated with ancestral traits.
Conclusion
The Co-ordinate Ascent Variational Inference scheme for approximate Bayesian inference on a generalised Phylogenetic Latent Variable Model provides estimates for the ancestral distribution of any collection of discrete and continuous traits.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.