README.md
In khughitt/smooth.ratio: Fast smoothing methods for R.
smooth.ratio
Window Size Independent Linear Time Smoothing for Biological Count Data
Note: This software was developed as part of a group project for a graduate level course at the University of Maryland, College Park (CMSC 702). The software was never formally published or peer-reviewed, so please use at your own risk.
Installation
You can use devtools to install smooth.ratio directly from Github:
library(devtools)
install_github('khughitt/smooth.ratio')
Usage Example: Whole Genome Bisulfite Sequencing
Overview
This example demonstrates the use of our smoothing approach for improving the signal-to-noise ratio for Whole Genome Bisulfite Sequencing (WGBS) methylation data.
WGBS data is inherently noisy, and as such the signatures of interesting features such as CpG islands is often obscured by numerous low coverage reads.
The data used for this analysis comes from a study by Hansen et al. (2011)\^1, and consists of three components:
- cpgsites - Genomic location (offset) of CpG sites in the dataset.
- methylation - Number of methylation reads at a given CpG site.
- coverage - Total number of reads at a given site.
The first variable (cpgsites) is a mx1 integer vector, while the later two variables are each mxn matrices, each with (n = 6) separate biological samples.
The smoothing method smooths the methylation values across time, weighting the contribution of each site by the ratio of the number of methylated reads to total reads at the site.
Analysis
First let's load sample data and take a look at what we have:
library(smoothratio)
load(file='data/sample.rda')
# Methylation reads at each site
head(methylation)
## [,1] [,2] [,3] [,4] [,5] [,6]
## [1,] 1 5 4 9 1 2
## [2,] 3 6 4 6 1 3
## [3,] 3 3 3 2 2 1
## [4,] 3 2 3 4 1 3
## [5,] 3 2 2 2 1 2
## [6,] 2 1 1 2 4 0
summary(methylation)
## V1 V2 V3 V4
## Min. : 0 Min. : 0 Min. : 0 Min. : 0
## 1st Qu.: 1 1st Qu.: 1 1st Qu.: 2 1st Qu.: 1
## Median : 4 Median : 3 Median : 4 Median : 3
## Mean : 4 Mean : 3 Mean : 4 Mean : 4
## 3rd Qu.: 6 3rd Qu.: 5 3rd Qu.: 6 3rd Qu.: 6
## Max. :16306 Max. :15501 Max. :16506 Max. :13777
## V5 V6
## Min. : 0 Min. : 0
## 1st Qu.: 1 1st Qu.: 1
## Median : 3 Median : 2
## Mean : 4 Mean : 3
## 3rd Qu.: 6 3rd Qu.: 5
## Max. :13826 Max. :14199
# Total number of reads at each site
head(coverage)
## [,1] [,2] [,3] [,4] [,5] [,6]
## [1,] 4 5 4 9 1 2
## [2,] 4 6 4 7 1 3
## [3,] 4 3 3 2 2 1
## [4,] 4 2 4 4 1 3
## [5,] 3 2 2 3 2 3
## [6,] 2 2 2 5 4 0
summary(coverage)
## V1 V2 V3 V4
## Min. : 0 Min. : 0 Min. : 0 Min. : 0
## 1st Qu.: 3 1st Qu.: 3 1st Qu.: 3 1st Qu.: 3
## Median : 5 Median : 5 Median : 6 Median : 6
## Mean : 6 Mean : 5 Mean : 6 Mean : 6
## 3rd Qu.: 8 3rd Qu.: 7 3rd Qu.: 8 3rd Qu.: 8
## Max. :17793 Max. :16594 Max. :18090 Max. :15064
## V5 V6
## Min. : 0 Min. : 0
## 1st Qu.: 3 1st Qu.: 3
## Median : 5 Median : 5
## Mean : 6 Mean : 5
## 3rd Qu.: 8 3rd Qu.: 7
## Max. :15222 Max. :16981
# Methylation locations (offset in Chromosome)
head(cpgsites)
## [,1]
## [1,] 10469
## [2,] 10471
## [3,] 10484
## [4,] 10489
## [5,] 10493
## [6,] 13079
range(cpgsites)
## [1] 10469 249239887
Let's now smooth a 20kb region of the chromosome.
indices = which((cpgsites > 830000) & (cpgsites < 850000))
result = smooth.ratio(cpgsites[indices], methylation[indices,], coverage[indices,], sigma_d=250)
# smoothed curve
head(result@fitted)
## [,1] [,2] [,3] [,4] [,5] [,6]
## [1,] 0.5574 0.6667 0.8601 0.7269 0.7871 0.5488
## [2,] 0.5566 0.6667 0.8592 0.7268 0.7858 0.5481
## [3,] 0.4445 0.6667 0.7191 0.7039 0.5656 0.4548
## [4,] 0.4280 0.6667 0.6780 0.7011 0.4993 0.4418
## [5,] 0.5491 0.0540 0.5256 0.4298 0.1003 0.6523
## [6,] 0.5496 0.0620 0.5294 0.4318 0.1151 0.6501
Finally, let's create some simple plots of our results.
plot(result, columns=1:2)
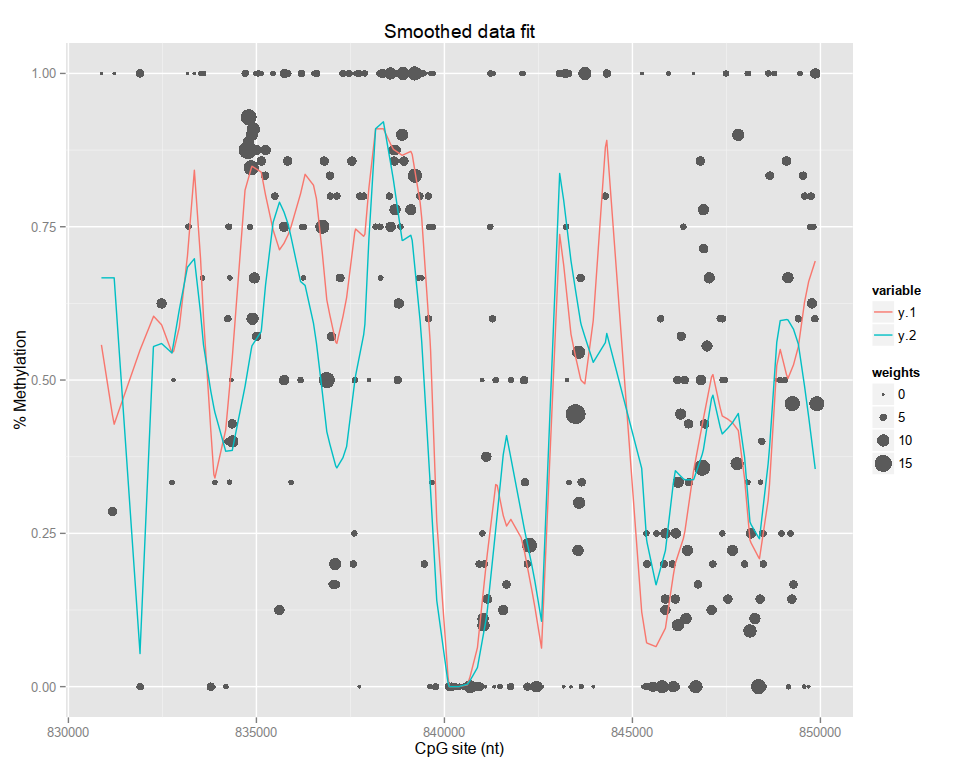
Done!
References
- Kasper Daniel Hansen, Winston Timp, Héctor Corrada Bravo, Sarven Sabunciyan, Benjamin Langmead, Oliver G McDonald, Bo Wen, Hao Wu, Yun Liu, Dinh Diep, Eirikur Briem, Kun Zhang, Rafael A Irizarry, Andrew P Feinberg, (2011) Increased Methylation Variation in Epigenetic Domains Across Cancer Types. Nature Genetics 43 10.1038/ng.865
khughitt/smooth.ratio documentation built on May 20, 2019, 9:23 a.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
smooth.ratio
Window Size Independent Linear Time Smoothing for Biological Count Data
Note: This software was developed as part of a group project for a graduate level course at the University of Maryland, College Park (CMSC 702). The software was never formally published or peer-reviewed, so please use at your own risk.
Installation
You can use devtools to install smooth.ratio directly from Github:
library(devtools)
install_github('khughitt/smooth.ratio')
Usage Example: Whole Genome Bisulfite Sequencing
Overview
This example demonstrates the use of our smoothing approach for improving the signal-to-noise ratio for Whole Genome Bisulfite Sequencing (WGBS) methylation data.
WGBS data is inherently noisy, and as such the signatures of interesting features such as CpG islands is often obscured by numerous low coverage reads.
The data used for this analysis comes from a study by Hansen et al. (2011)\^1, and consists of three components:
- cpgsites - Genomic location (offset) of CpG sites in the dataset.
- methylation - Number of methylation reads at a given CpG site.
- coverage - Total number of reads at a given site.
The first variable (cpgsites) is a mx1 integer vector, while the later two variables are each mxn matrices, each with (n = 6) separate biological samples.
The smoothing method smooths the methylation values across time, weighting the contribution of each site by the ratio of the number of methylated reads to total reads at the site.
Analysis
First let's load sample data and take a look at what we have:
library(smoothratio)
load(file='data/sample.rda')
# Methylation reads at each site
head(methylation)
## [,1] [,2] [,3] [,4] [,5] [,6]
## [1,] 1 5 4 9 1 2
## [2,] 3 6 4 6 1 3
## [3,] 3 3 3 2 2 1
## [4,] 3 2 3 4 1 3
## [5,] 3 2 2 2 1 2
## [6,] 2 1 1 2 4 0
summary(methylation)
## V1 V2 V3 V4
## Min. : 0 Min. : 0 Min. : 0 Min. : 0
## 1st Qu.: 1 1st Qu.: 1 1st Qu.: 2 1st Qu.: 1
## Median : 4 Median : 3 Median : 4 Median : 3
## Mean : 4 Mean : 3 Mean : 4 Mean : 4
## 3rd Qu.: 6 3rd Qu.: 5 3rd Qu.: 6 3rd Qu.: 6
## Max. :16306 Max. :15501 Max. :16506 Max. :13777
## V5 V6
## Min. : 0 Min. : 0
## 1st Qu.: 1 1st Qu.: 1
## Median : 3 Median : 2
## Mean : 4 Mean : 3
## 3rd Qu.: 6 3rd Qu.: 5
## Max. :13826 Max. :14199
# Total number of reads at each site
head(coverage)
## [,1] [,2] [,3] [,4] [,5] [,6]
## [1,] 4 5 4 9 1 2
## [2,] 4 6 4 7 1 3
## [3,] 4 3 3 2 2 1
## [4,] 4 2 4 4 1 3
## [5,] 3 2 2 3 2 3
## [6,] 2 2 2 5 4 0
summary(coverage)
## V1 V2 V3 V4
## Min. : 0 Min. : 0 Min. : 0 Min. : 0
## 1st Qu.: 3 1st Qu.: 3 1st Qu.: 3 1st Qu.: 3
## Median : 5 Median : 5 Median : 6 Median : 6
## Mean : 6 Mean : 5 Mean : 6 Mean : 6
## 3rd Qu.: 8 3rd Qu.: 7 3rd Qu.: 8 3rd Qu.: 8
## Max. :17793 Max. :16594 Max. :18090 Max. :15064
## V5 V6
## Min. : 0 Min. : 0
## 1st Qu.: 3 1st Qu.: 3
## Median : 5 Median : 5
## Mean : 6 Mean : 5
## 3rd Qu.: 8 3rd Qu.: 7
## Max. :15222 Max. :16981
# Methylation locations (offset in Chromosome)
head(cpgsites)
## [,1]
## [1,] 10469
## [2,] 10471
## [3,] 10484
## [4,] 10489
## [5,] 10493
## [6,] 13079
range(cpgsites)
## [1] 10469 249239887
Let's now smooth a 20kb region of the chromosome.
indices = which((cpgsites > 830000) & (cpgsites < 850000))
result = smooth.ratio(cpgsites[indices], methylation[indices,], coverage[indices,], sigma_d=250)
# smoothed curve
head(result@fitted)
## [,1] [,2] [,3] [,4] [,5] [,6]
## [1,] 0.5574 0.6667 0.8601 0.7269 0.7871 0.5488
## [2,] 0.5566 0.6667 0.8592 0.7268 0.7858 0.5481
## [3,] 0.4445 0.6667 0.7191 0.7039 0.5656 0.4548
## [4,] 0.4280 0.6667 0.6780 0.7011 0.4993 0.4418
## [5,] 0.5491 0.0540 0.5256 0.4298 0.1003 0.6523
## [6,] 0.5496 0.0620 0.5294 0.4318 0.1151 0.6501
Finally, let's create some simple plots of our results.
plot(result, columns=1:2)
Done!
References
- Kasper Daniel Hansen, Winston Timp, Héctor Corrada Bravo, Sarven Sabunciyan, Benjamin Langmead, Oliver G McDonald, Bo Wen, Hao Wu, Yun Liu, Dinh Diep, Eirikur Briem, Kun Zhang, Rafael A Irizarry, Andrew P Feinberg, (2011) Increased Methylation Variation in Epigenetic Domains Across Cancer Types. Nature Genetics 43 10.1038/ng.865
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.