In ningzhibin/rmdocpu: Metalab report upon provided templates
htmltools::img(src = "https://raw.githubusercontent.com/ningzhibin/rmdocpu/master/inst/rmd/iMetaReport.png",
alt = 'logo',
style = 'position:absolute; top:0; right:0; padding:10px;width:100px;height:100px;')
knitr::opts_chunk$set(echo = FALSE,warning = FALSE, message = FALSE, cache = FALSE)
# enviroment setup
source("https://raw.githubusercontent.com/ningzhibin/rmdocpu/master/inst/subfunctions_general.r")
source("https://raw.githubusercontent.com/ningzhibin/rmdocpu/master/inst/subfunctions_general_update.r")
library(tidyverse)
library(ggplot2)
library(d3heatmap)
library(plotly)
# other reqruied package:
# DT
#library(gplots)
# todo meta check
peptide.txt <- read.delim("peptides3.txt", header = TRUE,check.names = FALSE, stringsAsFactors = FALSE)
rmarkdown::render("MQ_report_peptides_indev.Rmd", params = list(input_datatable = peptide.txt))
rmarkdown::render("MQ_report_peptides.Rmd", params = list(input_datatable = your_readin_tbl, meta_table = meta_table_input))
#meta_table <- read.delim("metadata.txt", header = TRUE, check.names = FALSE, stringsAsFactors = FALSE) # test with meta file
tidy_peptides <- function(peptide.txt){
peptide_sequence <- peptide.txt$Sequence # only keep the first one
# do the row wise filtering
index_contaminant <- grep("\\+", peptide.txt$`Potential contaminant`) # note that + is a special character
index_reverse <- grep("\\+", peptide.txt$Reverse)
index_to_remove <- c(index_contaminant,index_reverse)
if(length(index_to_remove) >0){ # some times there are no rows to remove
peptide.txt <- peptide.txt[-index_to_remove,] # filtered table
peptide_sequence <- peptide_sequence[-index_to_remove] # filtered ids
}
n_contaminant <- length(index_contaminant)
n_reversed <- length(index_reverse)
# extra the intensity column matrix
if(any(grepl("LFQ intensity ", colnames(peptide.txt)))){ # if there are LFQ intensity columns, take out the LFQ columns
intensity_columns <- peptide.txt[,grep("LFQ intensity ", colnames(peptide.txt))]
colnames(intensity_columns) <- gsub("LFQ intensity ", "", colnames(intensity_columns))
}else{ # otherwise take out intensity column, even if there is one column, without any experiment desgin
intensity_columns <- peptide.txt[,grep("Intensity ", colnames(peptide.txt)),drop = FALSE]
colnames(intensity_columns)<-gsub("Intensity ", "", colnames(intensity_columns))
}
return(list("intensity_matrix" = intensity_columns,
"peptide_sequence" =peptide_sequence,
"n_contaminant" = n_contaminant,
"n_reversed" = n_reversed,
"score" = peptide.txt$Score,
"Charges" =peptide.txt$Charges,
"length" = peptide.txt$Length,
"misscleavage" = peptide.txt$"Missed cleavages"
))
}
# input
peptide.txt <- params$input_datatable
# Note: The folling analysis with meta info assumes that
# 1st columns as sample name, 2nd column as experiment name, 3rd column and after as grouping
meta_table <- params$meta_table
# tidy and process:
peptide_tidyed <- tidy_peptides(peptide.txt)
df_intensity <- peptide_tidyed$intensity_matrix
sparsity <- rowSums(df_intensity > 0) # here sparsity is number of present values
index_all_na_rows <- which(sparsity == 0)
df_intensity <- df_intensity[-index_all_na_rows,,drop = FALSE]
peptide_sequence <- peptide_tidyed$peptide_sequence[-index_all_na_rows]
# check meta_table
if( is.null(meta_table)){
meta_info <- "* **No meta information provided**"
}else if (any(is.na(meta_table))){
meta_info <- "* **The meta information provided has missing values, please check again**"
}else if(!(all(as.vector(colnames(df_intensity)) %in% meta_table[,2]) && all( meta_table[,2] %in% as.vector(colnames(df_intensity))))){
meta_info <- "* **The experiment in meta information provided do not match the experiment names in the peptide.txt, please check again**"
}else{
meta_info <- c("* **Groups: **",unique(meta_table[,3]))
}
# this figure height is for very tight figrues
figure_height <- 0.1*ncol(df_intensity)+4
Intro
This report provides some basic description of the peptide identificaiton from database search.
Users can use this to quickly check the overal quality of the experiment
Users can download the clean peptide quantification matrix for downstream analysis
Take-home figures
-
Number of contaminant: r peptide_tidyed$n_contaminant
-
Number of reversed: r peptide_tidyed$n_reversed
-
Number of qualified peptides: r nrow(peptide_tidyed$intensity_matrix)
-
Number of quailfied peptide without intensity information(0 intensitiy): r length(index_all_na_rows)
-
Number of experiment: r ncol(df_intensity)
-
All experiments: r colnames(df_intensity)
r meta_info
Peptide Charge States
Why charge state?
-
Peptide Charge distribution is a good sign of trypsin digestion and electric spray ionization.
-
In a typical ESI analysis of trytic digest, most of the peptides should have 2 charges, less peptides have 3 charges, because tryptic peptides have a lysine/arginie at the C-terminal, along with N-terminal contributing another charge. A possible miscleavage will contribute the third charge.
-
In a ESI procedure, peptides with 2 and more charges are easier to fragment and then identified by MS. However, too mnay charges will make the m/z of the peptide too small to escape the scan range, further more, it will also complicate the ms2 spectra.
-
if you see more peptides with charge 3 than charge 2 state,
-
It might indcate in-sufficient trypsin digesion, check the percentage of peptides with mis-cleavage site.
-
It migtht indcate the ESI is not sufficient/good enough.Check the distance between the ESI tip and MS oriface, if the ESI tip is dirty, if there is droplet occasionally.
peptide_charge <- as.data.frame(table(peptide_tidyed$Charges))
colnames(peptide_charge) = c("Charge_state", "Freq")
ggplot2::ggplot(data = peptide_charge)+
geom_col(aes(x = Charge_state,y = Freq))+
labs(title = "Charge Distribution", x = "Charge State",y = "Frequency") +
theme_bw() +
theme(plot.title = element_text(hjust = 0.5),
axis.text.x = element_text(angle = 90, hjust = 1),
panel.grid = element_blank())
Peptide Length
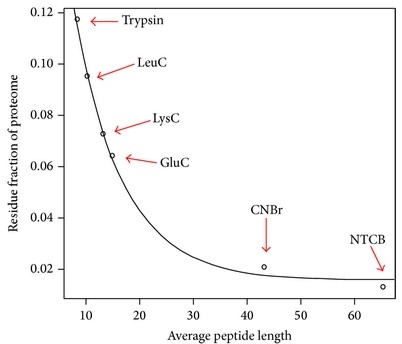
Why peptide length do you expect?
- Averge length of tryptic peptide is around 10.
- refer to this page for peptide length.
*
peptide_length <- as.data.frame(table(peptide_tidyed$length))
colnames(peptide_length) = c("peptide_length", "Freq")
ggplot2::ggplot(data = peptide_length)+
geom_col(aes(x = peptide_length,y = Freq))+
labs(title = "Peptide Length Distribution", x = "Peptide Length",y = "Frequency") +
theme_bw()+
theme(plot.title = element_text(hjust = 0.5),
axis.text.x = element_text(angle = 90, hjust = 1),
panel.grid = element_blank())
Peptide Score distribution
Why peptide length do you expect?
- The average score should be around 50
df_score <- data.frame(Score = peptide_tidyed$score)
ggplot2::ggplot(data=df_score, aes(Score)) +
geom_histogram(aes(y=..density..),color= "black",fill="white")+
geom_density(alpha=.2, fill="#5DADE2")+
labs(title = "Peptide Score Distribution", x = "Peptide Score",y = "Density") +
theme_bw()+
theme(plot.title = element_text(hjust = 0.5),
axis.text.x = element_text(angle = 90, hjust = 1),
panel.grid = element_blank())
Quantification Sparsity Distrubition
In most cases, label-free quantification provides a decent way for metaproteomics profiling.
Sparsity Profile
The more peptide of 100% presence the better
df_sparsity <- as.data.frame(table(sparsity))
df_sparsity_dec <- df_sparsity[order(df_sparsity$sparsity,decreasing = TRUE),]
df_sparsity_dec$sparsity <- factor(df_sparsity_dec$sparsity, levels = df_sparsity_dec$sparsity)
ggplot2::ggplot(data = df_sparsity_dec)+
geom_col(aes(x = sparsity,y = Freq))+
labs(title = "Sparsity Distribution", x = "Number of Presence",y = "Frequency") +
theme_bw()+
theme(plot.title = element_text(hjust = 0.5),
axis.text.x = element_text(angle = 90, hjust = 1),
panel.grid = element_blank())
Sparsity cummulative curve
Figure shows how many peptides have more than N presence, which helps to set the presence cutoff
#df_sparsity_dec <- df_sparsity[order(df_sparsity$sparsity,decreasing = TRUE),]
df_sparsity_dec$cumsum <- cumsum(df_sparsity_dec$Freq)
# redefine the sparisty sequence, in order to keep the order in the plot, This is the best way as far as I know
#df_sparsity_dec$sparsity <- factor(df_sparsity_dec$sparsity, levels = df_sparsity_dec$sparsity)
ggplot2::ggplot(data = df_sparsity_dec)+
geom_col(aes(x = sparsity,y = cumsum))+
labs(title = "Sparsity Distribution", x = "Number of Presence",y = "Frequency") +
theme_bw()+
theme(plot.title = element_text(hjust = 0.5),
axis.text.x = element_text(angle = 90, hjust = 1),
panel.grid = element_blank())
Peptide Intensity disitribution across samples/experiments
#note: in this log10 intenisty matrix, 0 is converted into infinity, therefore will not show in the box plot, and does not affect the distribution
df_intensity_log10 <-log10(df_intensity)
data_matrix_log10_melt<-reshape2::melt(as.matrix(df_intensity_log10))
colnames(data_matrix_log10_melt) <- c("Proteins", "Samples", "Log10(Inensity)")
ggplot(data_matrix_log10_melt)+
geom_boxplot(aes(x = Samples, y = `Log10(Inensity)`)) +
labs(title = "Peptide Intensity Distribution", x = "Number of Presence",y = "Frequency") +
theme_bw()+
theme(plot.title = element_text(hjust = 0.5),
axis.text.x = element_text(angle = 90, hjust = 1),
panel.grid = element_blank())+
coord_flip()
r if(ncol(df_intensity) > 1){ "# Overall Expression Profile"}
r if(ncol(df_intensity) > 1){ "## Heatmap"}
r if(ncol(df_intensity) > 1){"### Heatmap of Q100 peptides (with 100% presence across experiments)" }
# for files of big size, only show limited number of rows (Q100) for the test
# for interactive html display, only use top100 peptide
if(ncol(df_intensity) > 1){ # only if there are more than 1 columns in experssion matrix, we can do the heatmap and even cluster with 3 or more samples
sparsity <- rowSums(df_intensity > 0)
index_Q100 <- which(sparsity == ncol(df_intensity)) # index_Q100 is the index of Q100 peptide
df_intensity_Q100 <- df_intensity[index_Q100, , drop = FALSE] # the Q100 peptide matrix
# plot a heatmap of Q100
df_intensity_Q100_log10 <- log10(as.matrix(df_intensity_Q100))
matrix_quick_heatmap(df_intensity_Q100_log10, scale = "row")
#d3heatmap(df_intensity_Q100_log10,show_grid = FALSE) # this will slow down the rendering of the html rendering
}
# this is for the next section
if(length(index_Q100) >100){
topN = 100
}else{
topN = length(index_Q100)
}
r if(ncol(df_intensity) > 1){"### Heatmap of Top 100 Intensity peptides" }
if(ncol(df_intensity) > 1){ # only if there are more than 1 columns in experssion matrix
#heatmap of top N # will wrap this to a function
peptide_sequence_keep <- peptide_sequence[index_Q100] # keep the Q100 pepide sequence
df_intensity_Q100$peptide_total_intensity<- rowSums(df_intensity_Q100) # add total intensity column
df_intensity_Q100$peptide_sequence <- peptide_sequence_keep # add peptide sequnce column
df_intensity_topN <- dplyr::top_n(df_intensity_Q100, topN, peptide_total_intensity) # top 100 rows
rownames(df_intensity_topN) <- df_intensity_topN$peptide_sequence # give the row names back
df_intensity_topN <-df_intensity_topN[, 1: (ncol(df_intensity_topN)-2)] # delete the last 2 column
df_intensity_topN_log10 <-log10(df_intensity_topN) # log10
if(params$if_html){
d3heatmap(df_intensity_topN_log10,show_grid = FALSE) # this also works
# incorporate this figure is going to have 4M extra in file size,
#
# library(canvasXpress)
# canvasXpress(
# data=t(df_intensity_topN_log10),
# #smpAnnot=x,
# #varAnnot=z,
# heatmapIndicatorWidth = 200, # for the color bar
# heatmapIndicatorHeight = 30,
# heatmapIndicatorHistogram = TRUE,
#
# heatmapIndicatorPosition ="topLeft",
# colorSpectrum=list("blue", "white", "red"),
# #colorSpectrumBreaks =list(8, 9,10),
# graphType="Heatmap",
# heatmapCellBoxColor="rgb(255,255,255)",
# samplesClustered=TRUE,
# showTransition=FALSE,
# title="Expression Profile",
# variablesClustered=TRUE,
# smpLabelScaleFontFactor=0.5,
#
# width = 800,
# height = 800
# )
}else{
matrix_quick_heatmap(as.matrix(df_intensity_topN_log10), scale = "none")
}
}
r if(ncol(df_intensity) > 3){"# PCA Analysis"}
if(ncol(df_intensity) > 3){ # otherwise, no point doing PCA
#df_intensity_log10 <- log10(df_intensity+1)
if(is.null(meta_table)){
#pca <-PCA_wrapper_prcomp2(data_matrix = as.matrix(df_intensity_log10), inputation = TRUE)
PCA_result <- prcomp(t(df_intensity_Q100_log10))
loading <- as.data.frame(PCA_result$x)
# for screen plot
sd <- PCA_result$sde
var <- sd^2
var.percent <- var/sum(var) * 100
PCs <- paste("PC", 1:length(var.percent))
df_scree <- data.frame(PC = factor(PCs, levels = PCs), ratio =var.percent)
ggplot(data=df_scree, aes(x=PC, y=ratio)) + geom_bar(stat="identity")+
labs(title = "PCA Scree Plot", x = "Component", y = "Ratio of variance") +
theme_bw() +
theme(plot.title = element_text(hjust = 0.5),
axis.text.x = element_text(angle = 90, hjust = 1),
panel.grid = element_blank()) #+coord_flip()
if(params$if_html){
plot_ly(loading, x = ~PC1, y = ~PC2, z = ~PC3) %>%
add_markers() %>%
add_text(text = row.names(loading))
}else{
ggplot(loading, aes(x=PC1, y=PC2)) +geom_point(shape=1) +
labs(title = "PCA Plot", x = "PC1", y = "PC2") +
theme_bw() +
theme(plot.title = element_text(hjust = 0.5),
axis.text.x = element_text(angle = 90, hjust = 1),
panel.grid = element_blank())
library("scatterplot3d") # load
scatterplot3d(loading)
}
}else{
# do pca analysis with meta groupign information
pca <-PCA_wrapper_prcomp2(data_matrix = as.matrix(df_intensity_log10), data_meta = meta_table[,c(2,3)], inputation = FALSE)
# plotting
print(pca$pca_scree_plot)
cat("Scree plot ## shows the performance of the PCA analysis, the more percentage the top procomponets accounts, the better separation")
print(pca$pca_component_plot)
cat("2d PCA Component plot ##")
print(pca$pca_confidence)
cat("2d PCA Component plot ## with confidence boundaries")
print(pca$pca_component_plot_kmeans)
cat("2d PCA Component plot with K-means grouping, which is non-supervised grouping ")
# pca$pca_component_plot_3d_interactive
}
}
r if(length(unique(meta_table[,3])) >=2 && all(table(meta_table[,3]) >=2)) {"# ANOVA TEST \n**Peptides with significiant(p < 0.05) change between any groups**"}
# will do this block while: 1, more than (includeing) two meta/groups, 2, each group has more than (includeing) 2 samples.
if(length(unique(meta_table[,3])) >=2 && all(table(meta_table[,3]) >=2)){
pvalues_anova <- matrix_PostHoc(df_intensity,meta_table[,3])
df_intensity_p <- cbind(pvalues_anova, df_intensity)
rownames(df_intensity_p) <- peptide_sequence
df_intensity_p_filtered <- df_intensity_p[which(!is.na(df_intensity_p$p_PostHoc_pairs)),]
DT::datatable(df_intensity_p_filtered, extensions = 'Buttons',options = list(dom = "Blfrtip",scrollX=TRUE, buttons = list('copy', 'print', list(
extend = 'collection',
buttons = c('csv', 'excel', 'pdf'),
text = 'Download'
))))
}
Download the neat table
The table is clean peptides expression table, with reversed and contaminant removed.
The values are the LFQ(with label free quantification turned on) or raw protein intensity from Maxquant output.
You can download/export and start from this table for downstream analysis using the (i)Metalab faminly apps.
The talbe can be further visualized by our shiny apps shiny.imetalab.ca
df_intensity <- as.data.frame(df_intensity)
rownames(df_intensity) <- peptide_sequence
# DT table is not the bottle neck for slow loading, instead, plotly figures is
if(params$if_html){
DT::datatable(df_intensity[1:100,], extensions = 'Buttons',options = list(dom = "Blfrtip",scrollX=TRUE, buttons = list('copy', 'print', list(
extend = 'collection',
buttons = c('csv', 'excel', 'pdf'),
text = 'Download'
))))
}else{
knitr::kable(df_intensity[1:100,])
}
ningzhibin/rmdocpu documentation built on Feb. 3, 2022, 9:30 p.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
htmltools::img(src = "https://raw.githubusercontent.com/ningzhibin/rmdocpu/master/inst/rmd/iMetaReport.png", alt = 'logo', style = 'position:absolute; top:0; right:0; padding:10px;width:100px;height:100px;')
knitr::opts_chunk$set(echo = FALSE,warning = FALSE, message = FALSE, cache = FALSE) # enviroment setup source("https://raw.githubusercontent.com/ningzhibin/rmdocpu/master/inst/subfunctions_general.r") source("https://raw.githubusercontent.com/ningzhibin/rmdocpu/master/inst/subfunctions_general_update.r") library(tidyverse) library(ggplot2) library(d3heatmap) library(plotly) # other reqruied package: # DT #library(gplots) # todo meta check
peptide.txt <- read.delim("peptides3.txt", header = TRUE,check.names = FALSE, stringsAsFactors = FALSE) rmarkdown::render("MQ_report_peptides_indev.Rmd", params = list(input_datatable = peptide.txt)) rmarkdown::render("MQ_report_peptides.Rmd", params = list(input_datatable = your_readin_tbl, meta_table = meta_table_input)) #meta_table <- read.delim("metadata.txt", header = TRUE, check.names = FALSE, stringsAsFactors = FALSE) # test with meta file
tidy_peptides <- function(peptide.txt){ peptide_sequence <- peptide.txt$Sequence # only keep the first one # do the row wise filtering index_contaminant <- grep("\\+", peptide.txt$`Potential contaminant`) # note that + is a special character index_reverse <- grep("\\+", peptide.txt$Reverse) index_to_remove <- c(index_contaminant,index_reverse) if(length(index_to_remove) >0){ # some times there are no rows to remove peptide.txt <- peptide.txt[-index_to_remove,] # filtered table peptide_sequence <- peptide_sequence[-index_to_remove] # filtered ids } n_contaminant <- length(index_contaminant) n_reversed <- length(index_reverse) # extra the intensity column matrix if(any(grepl("LFQ intensity ", colnames(peptide.txt)))){ # if there are LFQ intensity columns, take out the LFQ columns intensity_columns <- peptide.txt[,grep("LFQ intensity ", colnames(peptide.txt))] colnames(intensity_columns) <- gsub("LFQ intensity ", "", colnames(intensity_columns)) }else{ # otherwise take out intensity column, even if there is one column, without any experiment desgin intensity_columns <- peptide.txt[,grep("Intensity ", colnames(peptide.txt)),drop = FALSE] colnames(intensity_columns)<-gsub("Intensity ", "", colnames(intensity_columns)) } return(list("intensity_matrix" = intensity_columns, "peptide_sequence" =peptide_sequence, "n_contaminant" = n_contaminant, "n_reversed" = n_reversed, "score" = peptide.txt$Score, "Charges" =peptide.txt$Charges, "length" = peptide.txt$Length, "misscleavage" = peptide.txt$"Missed cleavages" )) }
# input peptide.txt <- params$input_datatable # Note: The folling analysis with meta info assumes that # 1st columns as sample name, 2nd column as experiment name, 3rd column and after as grouping meta_table <- params$meta_table
# tidy and process: peptide_tidyed <- tidy_peptides(peptide.txt) df_intensity <- peptide_tidyed$intensity_matrix sparsity <- rowSums(df_intensity > 0) # here sparsity is number of present values index_all_na_rows <- which(sparsity == 0) df_intensity <- df_intensity[-index_all_na_rows,,drop = FALSE] peptide_sequence <- peptide_tidyed$peptide_sequence[-index_all_na_rows] # check meta_table if( is.null(meta_table)){ meta_info <- "* **No meta information provided**" }else if (any(is.na(meta_table))){ meta_info <- "* **The meta information provided has missing values, please check again**" }else if(!(all(as.vector(colnames(df_intensity)) %in% meta_table[,2]) && all( meta_table[,2] %in% as.vector(colnames(df_intensity))))){ meta_info <- "* **The experiment in meta information provided do not match the experiment names in the peptide.txt, please check again**" }else{ meta_info <- c("* **Groups: **",unique(meta_table[,3])) } # this figure height is for very tight figrues figure_height <- 0.1*ncol(df_intensity)+4
Intro
This report provides some basic description of the peptide identificaiton from database search. Users can use this to quickly check the overal quality of the experiment Users can download the clean peptide quantification matrix for downstream analysis
Take-home figures
-
Number of contaminant:
r peptide_tidyed$n_contaminant -
Number of reversed:
r peptide_tidyed$n_reversed -
Number of qualified peptides:
r nrow(peptide_tidyed$intensity_matrix) -
Number of quailfied peptide without intensity information(0 intensitiy):
r length(index_all_na_rows) -
Number of experiment:
r ncol(df_intensity) -
All experiments:
r colnames(df_intensity)
r meta_info
Peptide Charge States
Why charge state?
-
Peptide Charge distribution is a good sign of trypsin digestion and electric spray ionization.
-
In a typical ESI analysis of trytic digest, most of the peptides should have 2 charges, less peptides have 3 charges, because tryptic peptides have a lysine/arginie at the C-terminal, along with N-terminal contributing another charge. A possible miscleavage will contribute the third charge.
-
In a ESI procedure, peptides with 2 and more charges are easier to fragment and then identified by MS. However, too mnay charges will make the m/z of the peptide too small to escape the scan range, further more, it will also complicate the ms2 spectra.
-
if you see more peptides with charge 3 than charge 2 state,
-
It might indcate in-sufficient trypsin digesion, check the percentage of peptides with mis-cleavage site.
-
It migtht indcate the ESI is not sufficient/good enough.Check the distance between the ESI tip and MS oriface, if the ESI tip is dirty, if there is droplet occasionally.
peptide_charge <- as.data.frame(table(peptide_tidyed$Charges)) colnames(peptide_charge) = c("Charge_state", "Freq") ggplot2::ggplot(data = peptide_charge)+ geom_col(aes(x = Charge_state,y = Freq))+ labs(title = "Charge Distribution", x = "Charge State",y = "Frequency") + theme_bw() + theme(plot.title = element_text(hjust = 0.5), axis.text.x = element_text(angle = 90, hjust = 1), panel.grid = element_blank())
Peptide Length
Why peptide length do you expect?
- Averge length of tryptic peptide is around 10.
- refer to this page for peptide length.
*
peptide_length <- as.data.frame(table(peptide_tidyed$length)) colnames(peptide_length) = c("peptide_length", "Freq") ggplot2::ggplot(data = peptide_length)+ geom_col(aes(x = peptide_length,y = Freq))+ labs(title = "Peptide Length Distribution", x = "Peptide Length",y = "Frequency") + theme_bw()+ theme(plot.title = element_text(hjust = 0.5), axis.text.x = element_text(angle = 90, hjust = 1), panel.grid = element_blank())
Peptide Score distribution
Why peptide length do you expect?
- The average score should be around 50
df_score <- data.frame(Score = peptide_tidyed$score) ggplot2::ggplot(data=df_score, aes(Score)) + geom_histogram(aes(y=..density..),color= "black",fill="white")+ geom_density(alpha=.2, fill="#5DADE2")+ labs(title = "Peptide Score Distribution", x = "Peptide Score",y = "Density") + theme_bw()+ theme(plot.title = element_text(hjust = 0.5), axis.text.x = element_text(angle = 90, hjust = 1), panel.grid = element_blank())
Quantification Sparsity Distrubition
In most cases, label-free quantification provides a decent way for metaproteomics profiling.
Sparsity Profile
The more peptide of 100% presence the better
df_sparsity <- as.data.frame(table(sparsity)) df_sparsity_dec <- df_sparsity[order(df_sparsity$sparsity,decreasing = TRUE),] df_sparsity_dec$sparsity <- factor(df_sparsity_dec$sparsity, levels = df_sparsity_dec$sparsity) ggplot2::ggplot(data = df_sparsity_dec)+ geom_col(aes(x = sparsity,y = Freq))+ labs(title = "Sparsity Distribution", x = "Number of Presence",y = "Frequency") + theme_bw()+ theme(plot.title = element_text(hjust = 0.5), axis.text.x = element_text(angle = 90, hjust = 1), panel.grid = element_blank())
Sparsity cummulative curve
Figure shows how many peptides have more than N presence, which helps to set the presence cutoff
#df_sparsity_dec <- df_sparsity[order(df_sparsity$sparsity,decreasing = TRUE),] df_sparsity_dec$cumsum <- cumsum(df_sparsity_dec$Freq) # redefine the sparisty sequence, in order to keep the order in the plot, This is the best way as far as I know #df_sparsity_dec$sparsity <- factor(df_sparsity_dec$sparsity, levels = df_sparsity_dec$sparsity) ggplot2::ggplot(data = df_sparsity_dec)+ geom_col(aes(x = sparsity,y = cumsum))+ labs(title = "Sparsity Distribution", x = "Number of Presence",y = "Frequency") + theme_bw()+ theme(plot.title = element_text(hjust = 0.5), axis.text.x = element_text(angle = 90, hjust = 1), panel.grid = element_blank())
Peptide Intensity disitribution across samples/experiments
#note: in this log10 intenisty matrix, 0 is converted into infinity, therefore will not show in the box plot, and does not affect the distribution df_intensity_log10 <-log10(df_intensity) data_matrix_log10_melt<-reshape2::melt(as.matrix(df_intensity_log10)) colnames(data_matrix_log10_melt) <- c("Proteins", "Samples", "Log10(Inensity)") ggplot(data_matrix_log10_melt)+ geom_boxplot(aes(x = Samples, y = `Log10(Inensity)`)) + labs(title = "Peptide Intensity Distribution", x = "Number of Presence",y = "Frequency") + theme_bw()+ theme(plot.title = element_text(hjust = 0.5), axis.text.x = element_text(angle = 90, hjust = 1), panel.grid = element_blank())+ coord_flip()
r if(ncol(df_intensity) > 1){ "# Overall Expression Profile"}
r if(ncol(df_intensity) > 1){ "## Heatmap"}
r if(ncol(df_intensity) > 1){"### Heatmap of Q100 peptides (with 100% presence across experiments)" }
# for files of big size, only show limited number of rows (Q100) for the test # for interactive html display, only use top100 peptide if(ncol(df_intensity) > 1){ # only if there are more than 1 columns in experssion matrix, we can do the heatmap and even cluster with 3 or more samples sparsity <- rowSums(df_intensity > 0) index_Q100 <- which(sparsity == ncol(df_intensity)) # index_Q100 is the index of Q100 peptide df_intensity_Q100 <- df_intensity[index_Q100, , drop = FALSE] # the Q100 peptide matrix # plot a heatmap of Q100 df_intensity_Q100_log10 <- log10(as.matrix(df_intensity_Q100)) matrix_quick_heatmap(df_intensity_Q100_log10, scale = "row") #d3heatmap(df_intensity_Q100_log10,show_grid = FALSE) # this will slow down the rendering of the html rendering } # this is for the next section if(length(index_Q100) >100){ topN = 100 }else{ topN = length(index_Q100) }
r if(ncol(df_intensity) > 1){"### Heatmap of Top 100 Intensity peptides" }
if(ncol(df_intensity) > 1){ # only if there are more than 1 columns in experssion matrix #heatmap of top N # will wrap this to a function peptide_sequence_keep <- peptide_sequence[index_Q100] # keep the Q100 pepide sequence df_intensity_Q100$peptide_total_intensity<- rowSums(df_intensity_Q100) # add total intensity column df_intensity_Q100$peptide_sequence <- peptide_sequence_keep # add peptide sequnce column df_intensity_topN <- dplyr::top_n(df_intensity_Q100, topN, peptide_total_intensity) # top 100 rows rownames(df_intensity_topN) <- df_intensity_topN$peptide_sequence # give the row names back df_intensity_topN <-df_intensity_topN[, 1: (ncol(df_intensity_topN)-2)] # delete the last 2 column df_intensity_topN_log10 <-log10(df_intensity_topN) # log10 if(params$if_html){ d3heatmap(df_intensity_topN_log10,show_grid = FALSE) # this also works # incorporate this figure is going to have 4M extra in file size, # # library(canvasXpress) # canvasXpress( # data=t(df_intensity_topN_log10), # #smpAnnot=x, # #varAnnot=z, # heatmapIndicatorWidth = 200, # for the color bar # heatmapIndicatorHeight = 30, # heatmapIndicatorHistogram = TRUE, # # heatmapIndicatorPosition ="topLeft", # colorSpectrum=list("blue", "white", "red"), # #colorSpectrumBreaks =list(8, 9,10), # graphType="Heatmap", # heatmapCellBoxColor="rgb(255,255,255)", # samplesClustered=TRUE, # showTransition=FALSE, # title="Expression Profile", # variablesClustered=TRUE, # smpLabelScaleFontFactor=0.5, # # width = 800, # height = 800 # ) }else{ matrix_quick_heatmap(as.matrix(df_intensity_topN_log10), scale = "none") } }
r if(ncol(df_intensity) > 3){"# PCA Analysis"}
if(ncol(df_intensity) > 3){ # otherwise, no point doing PCA #df_intensity_log10 <- log10(df_intensity+1) if(is.null(meta_table)){ #pca <-PCA_wrapper_prcomp2(data_matrix = as.matrix(df_intensity_log10), inputation = TRUE) PCA_result <- prcomp(t(df_intensity_Q100_log10)) loading <- as.data.frame(PCA_result$x) # for screen plot sd <- PCA_result$sde var <- sd^2 var.percent <- var/sum(var) * 100 PCs <- paste("PC", 1:length(var.percent)) df_scree <- data.frame(PC = factor(PCs, levels = PCs), ratio =var.percent) ggplot(data=df_scree, aes(x=PC, y=ratio)) + geom_bar(stat="identity")+ labs(title = "PCA Scree Plot", x = "Component", y = "Ratio of variance") + theme_bw() + theme(plot.title = element_text(hjust = 0.5), axis.text.x = element_text(angle = 90, hjust = 1), panel.grid = element_blank()) #+coord_flip() if(params$if_html){ plot_ly(loading, x = ~PC1, y = ~PC2, z = ~PC3) %>% add_markers() %>% add_text(text = row.names(loading)) }else{ ggplot(loading, aes(x=PC1, y=PC2)) +geom_point(shape=1) + labs(title = "PCA Plot", x = "PC1", y = "PC2") + theme_bw() + theme(plot.title = element_text(hjust = 0.5), axis.text.x = element_text(angle = 90, hjust = 1), panel.grid = element_blank()) library("scatterplot3d") # load scatterplot3d(loading) } }else{ # do pca analysis with meta groupign information pca <-PCA_wrapper_prcomp2(data_matrix = as.matrix(df_intensity_log10), data_meta = meta_table[,c(2,3)], inputation = FALSE) # plotting print(pca$pca_scree_plot) cat("Scree plot ## shows the performance of the PCA analysis, the more percentage the top procomponets accounts, the better separation") print(pca$pca_component_plot) cat("2d PCA Component plot ##") print(pca$pca_confidence) cat("2d PCA Component plot ## with confidence boundaries") print(pca$pca_component_plot_kmeans) cat("2d PCA Component plot with K-means grouping, which is non-supervised grouping ") # pca$pca_component_plot_3d_interactive } }
r if(length(unique(meta_table[,3])) >=2 && all(table(meta_table[,3]) >=2)) {"# ANOVA TEST \n**Peptides with significiant(p < 0.05) change between any groups**"}
# will do this block while: 1, more than (includeing) two meta/groups, 2, each group has more than (includeing) 2 samples. if(length(unique(meta_table[,3])) >=2 && all(table(meta_table[,3]) >=2)){ pvalues_anova <- matrix_PostHoc(df_intensity,meta_table[,3]) df_intensity_p <- cbind(pvalues_anova, df_intensity) rownames(df_intensity_p) <- peptide_sequence df_intensity_p_filtered <- df_intensity_p[which(!is.na(df_intensity_p$p_PostHoc_pairs)),] DT::datatable(df_intensity_p_filtered, extensions = 'Buttons',options = list(dom = "Blfrtip",scrollX=TRUE, buttons = list('copy', 'print', list( extend = 'collection', buttons = c('csv', 'excel', 'pdf'), text = 'Download' )))) }
Download the neat table
The table is clean peptides expression table, with reversed and contaminant removed. The values are the LFQ(with label free quantification turned on) or raw protein intensity from Maxquant output. You can download/export and start from this table for downstream analysis using the (i)Metalab faminly apps.
The talbe can be further visualized by our shiny apps shiny.imetalab.ca
df_intensity <- as.data.frame(df_intensity) rownames(df_intensity) <- peptide_sequence # DT table is not the bottle neck for slow loading, instead, plotly figures is if(params$if_html){ DT::datatable(df_intensity[1:100,], extensions = 'Buttons',options = list(dom = "Blfrtip",scrollX=TRUE, buttons = list('copy', 'print', list( extend = 'collection', buttons = c('csv', 'excel', 'pdf'), text = 'Download' )))) }else{ knitr::kable(df_intensity[1:100,]) }
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.