Nothing
README.md
In MetaComp: EDGE Taxonomy Assignments Visualization
MetaComp
Metagenome taxonomy assignment comparison toolkit. The toolkit is being developed for EDGE platform and reflects its backend specificity. The routines, however, can be used as a stand-alone library for multi-project comparative visualization of taxonomy assignments obtained for metagenomic samples processed with GOTTCHA/GOTTCHA2, BWA, KRAKEN, METAPHLAN, DIAMOND, or PANGIA. The heatmaps can be also visualized with this D3.js-based code which allows to see the exact abundance values in each cell.






0.0 Installation from CRAN
install.packages("MetaComp")
to use the library, simply load it into R environment:
library(MetaComp)
0.1 Installation from latest sources
install.packages("devtools")
library(devtools)
install_github(repo = 'seninp-bioinfo/MetaComp')
1.0 Reading a single taxonomic assignment files
the_gottcha2_assignment <- load_edge_assignment(data_file_g2, type = 'gottcha2')
the_kraken_assignment <- load_edge_assignment(data_file_k, type = 'kraken')
the_pangia_assignment <- load_edge_assignment(data_file_p, type = 'pangia')
1.1 Reading multiple taxonomic assignment files
The package functions load_xxx_assignments (where xxx stands for gottcha, kraken, or metaphlan) are designed to read a tool-specific assignment files. The configuration file for these functions must be tab-delimeted two columns file where the first column is the project id (used as the project's name in plotting), and the second column is an actual assignment file path:
the_assignments_list_g2 <- load_edge_assignments(config_file_g2, type = 'gottcha2')
the_assignments_list_k <- load_edge_assignments(config_file_k, type = 'kraken')
the_assignments_list_p <- load_edge_assignments(config_file_pangia, type = 'pangia')
2.0 Merging multiple taxonomic assignments into a single table
The merge_edge_assignments function is capable to merge a named list of GOTTCHA, Kraken, or MetaPhlAn assignments into a single table using LEVEL and TAXA columns as ids.
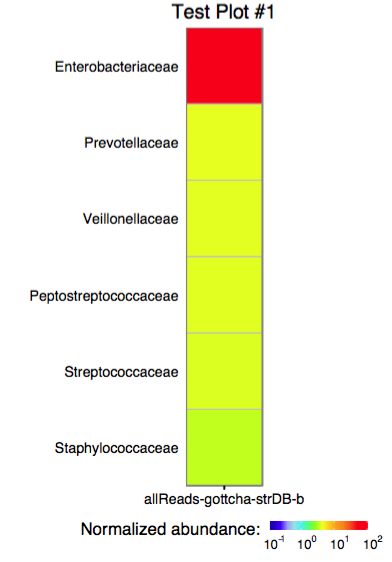
3.0 Plotting a single assignment as a heatmap
The function plot_edge_assignment accepts a single assignment table and outputs a ggplot object or produces a PDF plot using ggplot2's geom_tile.
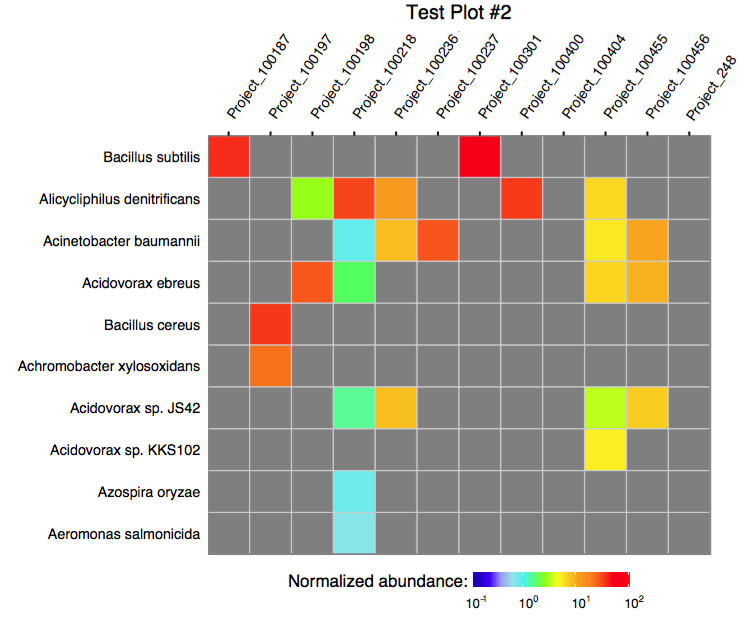
3.1 Plotting multiple assignments as a single heatmap
The function plot_merged_assignment accepts a single merged assignment table as an input and outputs a ggplot object or produces a PDF plot using ggplot2's geom_tile.
4.0. Running merge in a batch mode
The following script can be used to run the merge procedure in a batch mode:
# load library
require(MetaComp)
#
# configure runtime
options(echo = TRUE)
args <- commandArgs(trailingOnly = TRUE)
#
# print provided args
print(paste("provided args: ", args))
#
# acquire values
srcFile <- args[1]
destFile <- args[2]
taxonomyLevelArg <- args[3]
plotTitleArg <- args[4]
plotFileArg <- args[5]
#
# extended functionality was added in the release #3, and we don't want to break the legacy systems
#
if (length(args) > 5) {
rowLimitArg <- args[6]
sortingOrderArg <- args[7]
} else {
rowLimitArg <- 60
sortingOrderArg <- "abundance"
}
#
# read the data and produce the merged table
merged <- merge_edge_assignments(load_edge_assignments(srcFile, type = "gottcha2"))
#
# write the merge table as a TAB-delimeted file
write.table(merged, file = destFile, col.names = T, row.names = F, quote = T, sep = "\t")
#
# produce a PDF of the merged assignment
plot_merged_assignment(assignment = merged, taxonomy_level = taxonomyLevelArg,
sorting_order = sortingOrderArg, row_limit = base::strtoi(rowLimitArg),
plot_title = plotTitleArg, filename = plotFileArg)
To execute the scrip, use Rscript as shown below:
$> Rscript merge_and_plot_gottcha_assignments.R assignments_table_gottcha.txt merged_assignments.txt \
family "Merge test plot" merge_test 20 alphabetical
this command line arguments are (some of these are clickable -- so you can see examples):
Rscript - a way to execute the R script
merge_and_plot_gottcha_assignments.R- the above script filename
assignments_table_gottcha.txt - the tab delimeted table of assignments (two columns: project_id TAB assignment_path)
merged_assignments_gottcha.txt - the tab-delimeted output file name
family - a LEVEL at which the plot should be produced
"Merge test plot"- the output plot's title
merge_test - the output plot filename mask, ".pdf" and ".svg" files will be produced...
20 the max number of rows to plot (in the specified sorting order)
alphabetical the merged plot* sorting order
Try the MetaComp package in your browser
Any scripts or data that you put into this service are public.
MetaComp documentation built on May 2, 2019, 9:25 a.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
MetaComp
Metagenome taxonomy assignment comparison toolkit. The toolkit is being developed for EDGE platform and reflects its backend specificity. The routines, however, can be used as a stand-alone library for multi-project comparative visualization of taxonomy assignments obtained for metagenomic samples processed with GOTTCHA/GOTTCHA2, BWA, KRAKEN, METAPHLAN, DIAMOND, or PANGIA. The heatmaps can be also visualized with this D3.js-based code which allows to see the exact abundance values in each cell.
0.0 Installation from CRAN
install.packages("MetaComp")
to use the library, simply load it into R environment:
library(MetaComp)
0.1 Installation from latest sources
install.packages("devtools")
library(devtools)
install_github(repo = 'seninp-bioinfo/MetaComp')
1.0 Reading a single taxonomic assignment files
the_gottcha2_assignment <- load_edge_assignment(data_file_g2, type = 'gottcha2')
the_kraken_assignment <- load_edge_assignment(data_file_k, type = 'kraken')
the_pangia_assignment <- load_edge_assignment(data_file_p, type = 'pangia')
1.1 Reading multiple taxonomic assignment files
The package functions load_xxx_assignments (where xxx stands for gottcha, kraken, or metaphlan) are designed to read a tool-specific assignment files. The configuration file for these functions must be tab-delimeted two columns file where the first column is the project id (used as the project's name in plotting), and the second column is an actual assignment file path:
the_assignments_list_g2 <- load_edge_assignments(config_file_g2, type = 'gottcha2')
the_assignments_list_k <- load_edge_assignments(config_file_k, type = 'kraken')
the_assignments_list_p <- load_edge_assignments(config_file_pangia, type = 'pangia')
2.0 Merging multiple taxonomic assignments into a single table
The merge_edge_assignments function is capable to merge a named list of GOTTCHA, Kraken, or MetaPhlAn assignments into a single table using LEVEL and TAXA columns as ids.
3.0 Plotting a single assignment as a heatmap
The function plot_edge_assignment accepts a single assignment table and outputs a ggplot object or produces a PDF plot using ggplot2's geom_tile.
3.1 Plotting multiple assignments as a single heatmap
The function plot_merged_assignment accepts a single merged assignment table as an input and outputs a ggplot object or produces a PDF plot using ggplot2's geom_tile.
4.0. Running merge in a batch mode
The following script can be used to run the merge procedure in a batch mode:
# load library
require(MetaComp)
#
# configure runtime
options(echo = TRUE)
args <- commandArgs(trailingOnly = TRUE)
#
# print provided args
print(paste("provided args: ", args))
#
# acquire values
srcFile <- args[1]
destFile <- args[2]
taxonomyLevelArg <- args[3]
plotTitleArg <- args[4]
plotFileArg <- args[5]
#
# extended functionality was added in the release #3, and we don't want to break the legacy systems
#
if (length(args) > 5) {
rowLimitArg <- args[6]
sortingOrderArg <- args[7]
} else {
rowLimitArg <- 60
sortingOrderArg <- "abundance"
}
#
# read the data and produce the merged table
merged <- merge_edge_assignments(load_edge_assignments(srcFile, type = "gottcha2"))
#
# write the merge table as a TAB-delimeted file
write.table(merged, file = destFile, col.names = T, row.names = F, quote = T, sep = "\t")
#
# produce a PDF of the merged assignment
plot_merged_assignment(assignment = merged, taxonomy_level = taxonomyLevelArg,
sorting_order = sortingOrderArg, row_limit = base::strtoi(rowLimitArg),
plot_title = plotTitleArg, filename = plotFileArg)
To execute the scrip, use Rscript as shown below:
$> Rscript merge_and_plot_gottcha_assignments.R assignments_table_gottcha.txt merged_assignments.txt \
family "Merge test plot" merge_test 20 alphabetical
this command line arguments are (some of these are clickable -- so you can see examples):
Rscript - a way to execute the R script
merge_and_plot_gottcha_assignments.R- the above script filename
assignments_table_gottcha.txt - the tab delimeted table of assignments (two columns: project_id TAB assignment_path)
merged_assignments_gottcha.txt - the tab-delimeted output file name
family - a LEVEL at which the plot should be produced
"Merge test plot"- the output plot's title
merge_test - the output plot filename mask, ".pdf" and ".svg" files will be produced...
20 the max number of rows to plot (in the specified sorting order)
alphabetical the merged plot* sorting order
Try the MetaComp package in your browser
Any scripts or data that you put into this service are public.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
{kind=link}
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.