Nothing
README.md
In genekitr: Gene Analysis Toolkit






Overview
Genekitr is a gene analysis toolkit based on R.
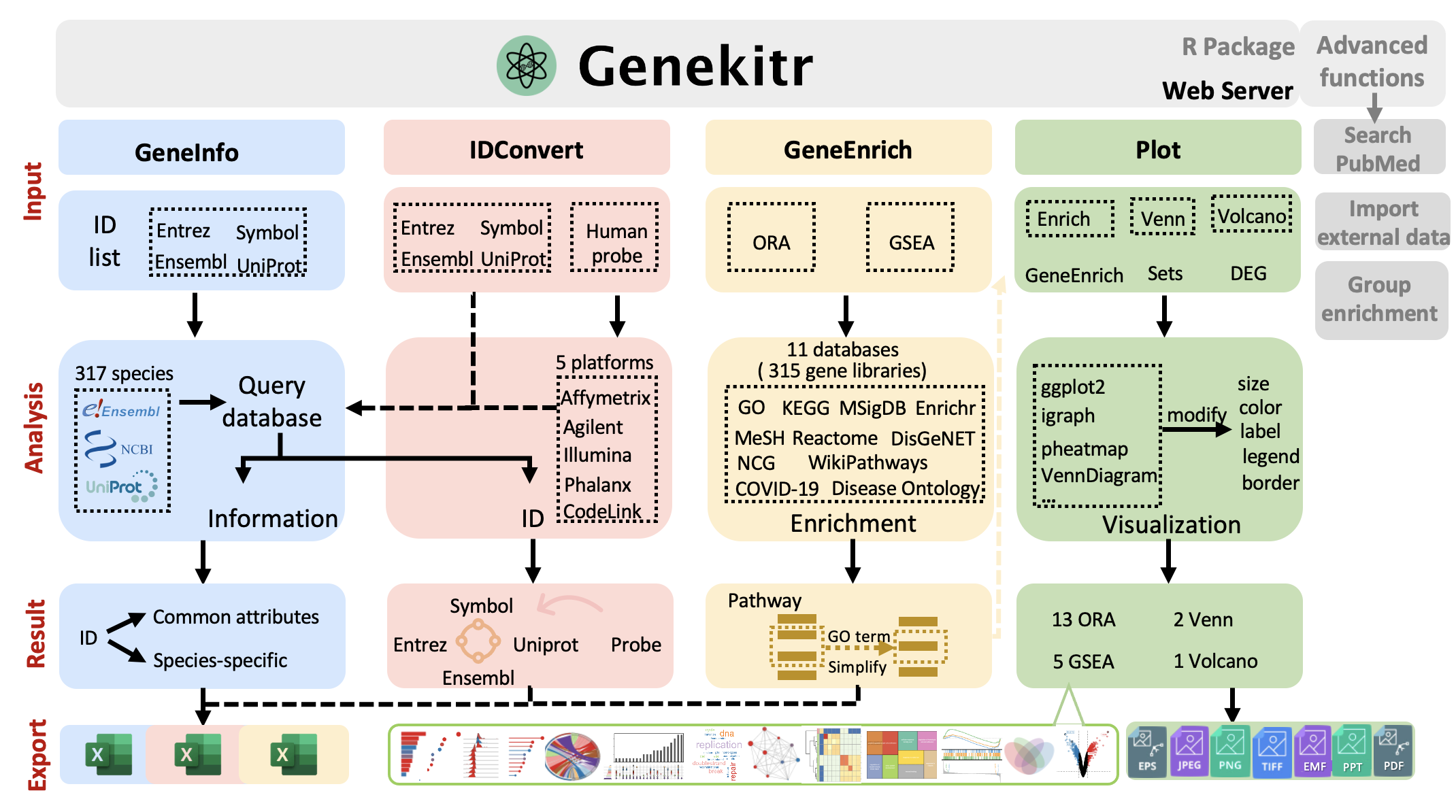
Five core features:
- Search: gene-related information (exp. gene functional summary, gene name, location, GC content, gene biotype ...) and PubMed records
-
Convert: ID conversion among Symbol & Alias, NCBI Entrez, Ensembl ,Uniprot and human microarray probe
-
Analysis: users could select interested gene set from hundreds of gene sets for both model and non-model species, including GO (BP, CC and MF), KEGG (pathway, module, enzyme, network, drug and disease), WikiPathways, MSigDB, EnrichrDB, Reactome, MeSH, DisGeNET, Disease Ontology (DO), Network of Cancer Gene (NCG) (version 6 and v7) and COVID-19. Gene enrichment analysis (GSA) contains both over representation analysis (ORA) and gene set enrichment analysis (GSEA) methods. ORA is capable of supporting multi-group comparisons.
-
Plot: easily generate 13 ORA plots, 5 GSEA plots, 2 Venn plots, and 1 Volcano plot with customizable features such as text, color, border, axis, and legend. The function is capable of accepting a dataframe as input and supports GeneOntology website results based on PantherDB..
- Export: quickly export numerous datasets as different sheets within a single Excel file.
Supported organisms:
For more details, please refer to this site.
- Search & ID conversion: 195 vertebrate species, 120 plant species and 2 bacteria species
- Enrichment analysis: GO supports 143 species, KEGG supports 8213 species, MeSH supports 71 species, MsigDb supports 20 species, WikiPahtwaysupports 16 species, Reactome supports 11 species, EnrichrDB supports 5 species and human-specific gene sets (DO, NCG, DisGeNET and COVID-19)
🛠 Installation
```R
check current version
packageVersion('genekitr')
```
Install stable version from CRAN:
install.packages("genekitr")
Install development version from GitHub:
remotes::install_github("GangLiLab/genekitr")
Install development version from Gitee (for CHN mainland users):
remotes::install_git("https://gitee.com/genekitr/pacakge_genekitr")
📚 Vignette
ENGLISH: https://www.genekitr.fun/
🧙🏻♂️ Tell a story ~ why develop genekitr?
Genes, the essence of life's art,
Omics research's fundamental part,
Like cells in our physical frame,
Their study reveals life's vibrant flame.
Let me tell you a story about Mr. Doodle, a computational biology student working with his PI.
Scene 1: repeat work
One day, PI gave him 30 genes to check for their locations and exact names, preferably with sequences.
Mr. Doodle searched for each gene on NCBI, copying and pasting the information into an Excel sheet. He sent the file to PI an hour later, and received praise for his work. But just when he thought he was done, PI gave him another 50 genes to check!
Despite feeling a little overwhelmed, Mr. Doodle repeated the same process with determination, determined to complete the task to the best of his abilities.
Doodle wondered how to avoid having to repeatedly search for the same information.?
Scene 2: embarrassing name
Once upon a time, PI gave Mr. Doodle a DEG matrix and a target gene list file. The task was to determine if the target gene was up-regulated after treatment.
Mr. Doodle searched the matrix but couldn't find the PDL1 gene, even though it was in the gene list. He asked PI about it, and PI explained that the gene was listed as CD274, which is an alias for PDL1.
This left Mr. Doodle feeling a little confused. He wondered how to distinguish between real gene names and aliases.
Doodle wondered how to differentiate between a real gene name and an alias.
Scene 3: outdated database
Mr. Doodle was analyzing KEGG pathways for the up-regulated genes in the last DEG matrix. However, KEGG only supported Entrez IDs, and the genes were listed by their symbols.
Mr. Doodle needed to convert the gene symbols to Entrez IDs, but he found that some symbols did not match the corresponding Entrez IDs. However, he discovered that NCBI had the correct IDs.
Mr. Doodle realized that he was using an outdated org.Hs.eg.db v3.15 annotation package. After updating to the current version, v3.17, he was finally able to obtain all the matched IDs, and continue his analysis of the KEGG pathways.
Doodle wondered if there was a method to help him obtain updated results automatically, instead of having to check them manually every time.
Scene 4: imcompatible format
PI did some fancy enrichment analysis all by himself on a website called GeneOntology. He then asked Mr.Doodle to help him make a pretty picture of the results. . "Can you make a bubble plot for me and show the FoldEnrichment on the x-axis?" he asked with a smile. Doodle tried to use a fancy R package called clusterProfiler, but it wouldn't work with the data. So, he bravely coded it himself using ggplot2.
Doodle wondered why there isn't a tool that supports easy data frames.
Scene 5: annoying plot theme
Doodle finally finished making the bubble plot and sent it to PI. After 15 minutes, PI sent him a message with a smile: "the text is too small, and can you make the background white with a border size of 4 points?" Doodle tweaked the ggplot theme and made the changes in 10 minutes. But, after a little while, PI sent another message saying, "The border is too thick in the second version. Can you please redo it?"
Doodle wondered if there was a function that could help him process the plot theme instead of having to modify the current code repeatedly.
Scene 6: limited plot types
PI gave Doodle the GO enrichment analysis result and asked him to think of a creative way to display it. Doodle found that each tool had its specific plot. For example, WEGO could compare BP, CC, and MF terms; GOplot had a chord plot to show the relationship between genes and GO terms; and clusterProfilersupported enriched map and network, which could explore the relationship among enriched terms. However, there was a big problem - the input data for each tool was not compatible, making it inconvenient to plot WEGO plots using clusterProfiler objects.
Doodle wondered if there was a method that could produce beautiful plots from different tools using a universal data format.
Scene 7: chaotic export files
Doodle finished conducting differential expression analysis and GO/KEGG enrichment analysis. PI asked him to send over all the result files. Doodle saved the results into three separate excel files, naming them "DEG_data.xlsx," "GO_enrich.xlsx," and "KEGG_enrich.xlsx." He then compressed the three files into a zipped folder, naming it after the date, and sent it to PI. After a while, PI asked him if he could put all three results into a single excel file.
Doodle wondered if there is a way to save all data into a single file without having to perform many manual operations.?
If you have encountered similar problems like Mr. Doodle, give genekitr a try!
✍️ Author

🔖 Citation
For now, the paper is published. Please cite:
Liu, Y., Li, G. Empowering biologists to decode omics data: the Genekitr R package and web server. BMC Bioinformatics 24, 214 (2023). https://doi.org/10.1186/s12859-023-05342-9
💓 Welcome to contribute
If you are interested in genekitr, welcome contribute your ideas as follows:
- Git clone this project
- Double click
genekitr.Rproj to open RStudio
- Modify source code in
R/ folder
- Run
devtools::check() to make sure no errors, warnings or notes
- Pull request and describe clearly your changes
Try the genekitr package in your browser
Any scripts or data that you put into this service are public.
genekitr documentation built on Sept. 11, 2024, 9:10 p.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
![]()
![]()
Overview
Genekitr is a gene analysis toolkit based on R.
Five core features:
- Search: gene-related information (exp. gene functional summary, gene name, location, GC content, gene biotype ...) and PubMed records
-
Convert: ID conversion among
Symbol & Alias,NCBI Entrez,Ensembl,Uniprotandhuman microarray probe -
Analysis: users could select interested gene set from hundreds of gene sets for both model and non-model species, including GO (BP, CC and MF), KEGG (pathway, module, enzyme, network, drug and disease), WikiPathways, MSigDB, EnrichrDB, Reactome, MeSH, DisGeNET, Disease Ontology (DO), Network of Cancer Gene (NCG) (version 6 and v7) and COVID-19. Gene enrichment analysis (GSA) contains both over representation analysis (ORA) and gene set enrichment analysis (GSEA) methods. ORA is capable of supporting multi-group comparisons.
-
Plot: easily generate 13 ORA plots, 5 GSEA plots, 2 Venn plots, and 1 Volcano plot with customizable features such as text, color, border, axis, and legend. The function is capable of accepting a dataframe as input and supports GeneOntology website results based on PantherDB..
- Export: quickly export numerous datasets as different sheets within a single Excel file.
Supported organisms:
For more details, please refer to this site.
- Search & ID conversion: 195 vertebrate species, 120 plant species and 2 bacteria species
- Enrichment analysis: GO supports 143 species, KEGG supports 8213 species, MeSH supports 71 species, MsigDb supports 20 species, WikiPahtwaysupports 16 species, Reactome supports 11 species, EnrichrDB supports 5 species and human-specific gene sets (DO, NCG, DisGeNET and COVID-19)
🛠 Installation
```R
check current version
packageVersion('genekitr') ```
Install stable version from CRAN:
install.packages("genekitr")
Install development version from GitHub:
remotes::install_github("GangLiLab/genekitr")
Install development version from Gitee (for CHN mainland users):
remotes::install_git("https://gitee.com/genekitr/pacakge_genekitr")
📚 Vignette
ENGLISH: https://www.genekitr.fun/
🧙🏻♂️ Tell a story ~ why develop genekitr?
Genes, the essence of life's art, Omics research's fundamental part,
Like cells in our physical frame, Their study reveals life's vibrant flame.
Let me tell you a story about Mr. Doodle, a computational biology student working with his PI.
Scene 1: repeat work
One day, PI gave him 30 genes to check for their locations and exact names, preferably with sequences.
Mr. Doodle searched for each gene on NCBI, copying and pasting the information into an Excel sheet. He sent the file to PI an hour later, and received praise for his work. But just when he thought he was done, PI gave him another 50 genes to check!
Despite feeling a little overwhelmed, Mr. Doodle repeated the same process with determination, determined to complete the task to the best of his abilities.
Doodle wondered how to avoid having to repeatedly search for the same information.?
Scene 2: embarrassing name
Once upon a time, PI gave Mr. Doodle a DEG matrix and a target gene list file. The task was to determine if the target gene was up-regulated after treatment.
Mr. Doodle searched the matrix but couldn't find the PDL1 gene, even though it was in the gene list. He asked PI about it, and PI explained that the gene was listed as CD274, which is an alias for PDL1.
This left Mr. Doodle feeling a little confused. He wondered how to distinguish between real gene names and aliases.
Doodle wondered how to differentiate between a real gene name and an alias.
Scene 3: outdated database
Mr. Doodle was analyzing KEGG pathways for the up-regulated genes in the last DEG matrix. However, KEGG only supported Entrez IDs, and the genes were listed by their symbols.
Mr. Doodle needed to convert the gene symbols to Entrez IDs, but he found that some symbols did not match the corresponding Entrez IDs. However, he discovered that NCBI had the correct IDs.
Mr. Doodle realized that he was using an outdated org.Hs.eg.db v3.15 annotation package. After updating to the current version, v3.17, he was finally able to obtain all the matched IDs, and continue his analysis of the KEGG pathways.
Doodle wondered if there was a method to help him obtain updated results automatically, instead of having to check them manually every time.
Scene 4: imcompatible format
PI did some fancy enrichment analysis all by himself on a website called GeneOntology. He then asked Mr.Doodle to help him make a pretty picture of the results. . "Can you make a bubble plot for me and show the FoldEnrichment on the x-axis?" he asked with a smile. Doodle tried to use a fancy R package called clusterProfiler, but it wouldn't work with the data. So, he bravely coded it himself using ggplot2.
Doodle wondered why there isn't a tool that supports easy data frames.
Scene 5: annoying plot theme
Doodle finally finished making the bubble plot and sent it to PI. After 15 minutes, PI sent him a message with a smile: "the text is too small, and can you make the background white with a border size of 4 points?" Doodle tweaked the ggplot theme and made the changes in 10 minutes. But, after a little while, PI sent another message saying, "The border is too thick in the second version. Can you please redo it?"
Doodle wondered if there was a function that could help him process the plot theme instead of having to modify the current code repeatedly.
Scene 6: limited plot types
PI gave Doodle the GO enrichment analysis result and asked him to think of a creative way to display it. Doodle found that each tool had its specific plot. For example, WEGO could compare BP, CC, and MF terms; GOplot had a chord plot to show the relationship between genes and GO terms; and clusterProfilersupported enriched map and network, which could explore the relationship among enriched terms. However, there was a big problem - the input data for each tool was not compatible, making it inconvenient to plot WEGO plots using clusterProfiler objects.
Doodle wondered if there was a method that could produce beautiful plots from different tools using a universal data format.
Scene 7: chaotic export files
Doodle finished conducting differential expression analysis and GO/KEGG enrichment analysis. PI asked him to send over all the result files. Doodle saved the results into three separate excel files, naming them "DEG_data.xlsx," "GO_enrich.xlsx," and "KEGG_enrich.xlsx." He then compressed the three files into a zipped folder, naming it after the date, and sent it to PI. After a while, PI asked him if he could put all three results into a single excel file.
Doodle wondered if there is a way to save all data into a single file without having to perform many manual operations.?
If you have encountered similar problems like Mr. Doodle, give genekitr a try!
✍️ Author

🔖 Citation
For now, the paper is published. Please cite:
Liu, Y., Li, G. Empowering biologists to decode omics data: the Genekitr R package and web server. BMC Bioinformatics 24, 214 (2023). https://doi.org/10.1186/s12859-023-05342-9
💓 Welcome to contribute
If you are interested in genekitr, welcome contribute your ideas as follows:
- Git clone this project
- Double click
genekitr.Rprojto open RStudio - Modify source code in
R/folder - Run
devtools::check()to make sure no errors, warnings or notes - Pull request and describe clearly your changes
Try the genekitr package in your browser
Any scripts or data that you put into this service are public.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.