paper/paper.md
In AntonelliLab/restez: Create and Query a Local Copy of 'GenBank' in R
title: 'restez: Create and Query a Local Copy of GenBank in R'
tags:
- GenBank
- nucleotides
- R
- DNA
- sequence
- NCBI
- rstats
authors:
- name: Dominic J. Bennett
orcid: 0000-0003-2722-1359
affiliation: "1, 2"
- name: Hannes Hettling
affiliation: "3"
- name: Daniele Silvestro
orcid: 0000-0003-0100-0961
affiliation: "1, 2"
- name: Rutger Vos
orcid: 0000-0001-9254-7318
affiliation: "3"
- name: Alexandre Antonelli
orcid: 0000-0003-1842-9297
affiliation: "1, 2, 4"
affiliations:
- name: Gothenburg Global Biodiversity Centre, Box 461, SE-405 30 Gothenburg, Sweden
index: 1
- name: Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, SE-405 30 Gothenburg, Sweden
index: 2
- name: Naturalis Biodiversity Center, P.O. Box 9517, 2300 RA Leiden, The Netherlands
index: 3
- name: Gothenburg Botanical Garden, SE 41319 Gothenburg, Sweden
index: 4
date: 27 November 2018
bibliography: paper.bib
Summary
Downloading sequences and sequence information from GenBank [@Benson2013] and related NCBI databases is often performed via the NCBI API, Entrez [@Ostell2002]. Entrez, however, has a limit on the number of requests, thus downloading large amounts of sequence data in this way can be inefficient. For situations where a large number of Entrez calls is made, downloading may take days, weeks or even months and could result in a user’s IP address being blacklisted from the NCBI services due to server overload. Additionally, Entrez limits the number of entries that can be retrieved at once, requiring a user to develop code for querying in batches.
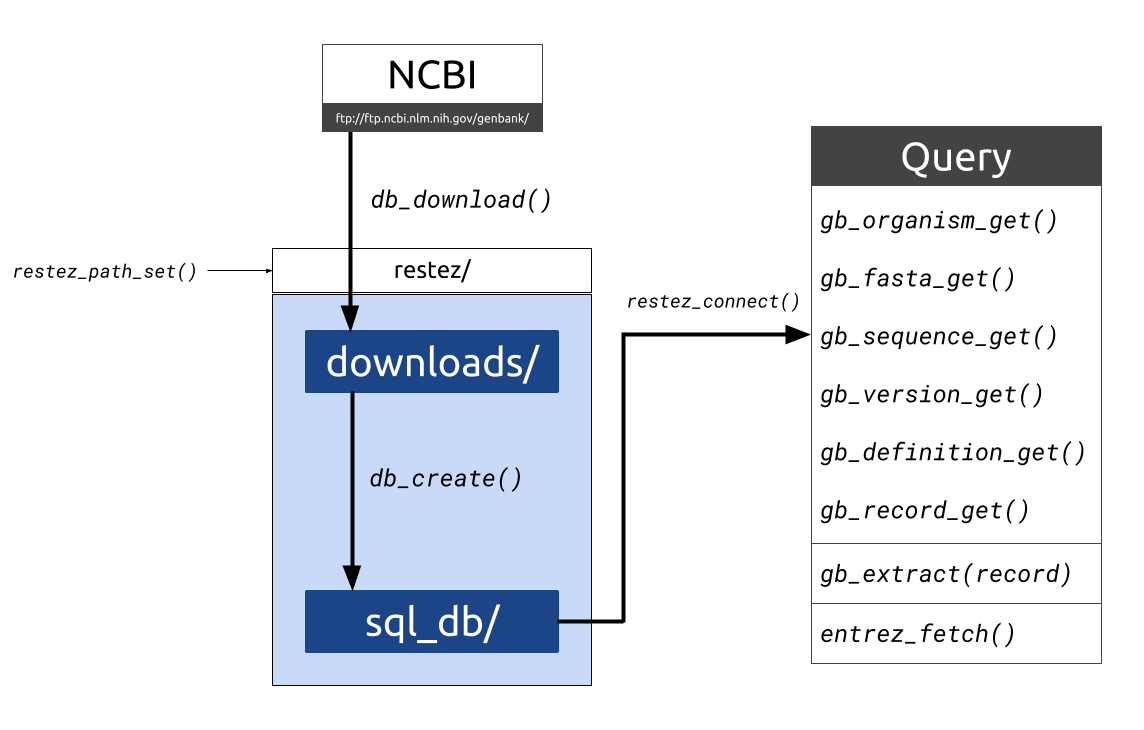
The restez package [@restez_z] aims to make sequence retrieval more efficient by allowing a user to download the GenBank database, either in its entirety or in subsets, to their local machine and query this local database instead. This process is more time efficient as GenBank downloads are made via NCBI’s FTP server using compressed sequence files. With a good internet connection and a computer with currently standard capabilities, a database comprising 7 GB of sequence information (i.e. the total sequence data available for Rodentia as of 27 June 2018) can be generated in less than 10 minutes. (For an outline of the functions and structure of restez, see Figure 1.)
Rentrez integration
rentrez [@Winter2017] is a popular R package for querying NCBI’s databases via Entrez in R. To maximize the compatibility of restez, we implemented wrapper functions with the same names and arguments as the rentrez equivalents. Whenever a wrapper function is called the local database copy is searched first. If IDs are missing in the local database a secondary call to Entrez is made via the internet. This allows for easy employment of restez in scripts and packages that are already using rentrez. At a minimum, a user currently using rentrez will only need to create a local subset of the GenBank database, call restez instead of rentrez and ensure the restez database is connected.
Examples
A small example
After a restez database has been set-up, we can retrieve all the sequences from an rentrez::entrez_search() with a single command.
# Use rentrez to search for accession IDs of interest
# Sequences in fasta format can then be retrieved with entrez_fetch
res <- rentrez::entrez_fetch(db = 'nuccore', id = ids, rettype = 'fasta')
# ^ likely to raise an error if too many IDs
res <- restez::entrez_fetch(db = 'nuccore', id = ids, rettype = 'fasta')
# ^ not likely to raise an error
A large example
phylotaR is an R package for retrieving and identifying orthologous sequence clusters from GenBank as a first step in a phylogenetic analysis [@Bennett2018]. Because the package runs an automated pipeline, multiple queries to GenBank via Entrez are made using the rentrez package. As a result, for large taxonomic groups containing well-sequenced organisms the pipeline can take a long time to complete.
library(phylotaR)
# run phylotaR pipeline for New World Monkeys
txid <- 9479 # taxonomic ID
setup(wd = 'nw_monkeys', txid = txid)
run(wd = wd)
# ^ takes around 40 minutes
We can download and create a local copy of the primates GenBank locally and re-run the above code with a library call to restez for speed-up gains and increased code reliability.
# setup database
library(restez)
# Specify path to a local directory in which database will be stored
# Make sure you have sufficient disk space!
restez_path_set(filepath = 'restez_db')
db_download(db = 'nucleotide') # Interactively download GenBank data
db_create(db = 'nucleotide')
Now when re-running the first phylotaR code block with the inclusion of the restez package, the procedure completes approximately eight times faster.
# run phylotaR again
library(phylotaR)
library(restez)
restez_path_set(filepath = 'restez_db')
txid <- 9479
setup(wd = 'nw_monkeys', txid = txid)
run(wd = wd)
# ^ takes around 5 minutes
For more detailed and up-to-date examples and tutorials, see the restez GitHub page [@restez_gh].
Availability
restez is open source software made available under the MIT license. It can be installed through CRAN [@restez_cran], install.package("restez"), or from its GitHub source code repository using the devtools package, e.g. as follows: devtools::install_github("ropensci/restez")
Funding
This package has been developed as part of the supersmartR project [@supersmartR] which has received funding through A.A. (from the Swedish Research Council [B0569601], the Swedish Foundation for Strategic Research, a Wallenberg Academy Fellowship, the Faculty of Sciences at the University of Gothenburg, the Wenner-Gren Foundations, and the David Rockefeller Center for Latin American Studies at Harvard University) and through D.S. (from the Swedish Research Council [2015-04748]).
References
AntonelliLab/restez documentation built on April 22, 2024, 7:27 p.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
title: 'restez: Create and Query a Local Copy of GenBank in R' tags: - GenBank - nucleotides - R - DNA - sequence - NCBI - rstats authors: - name: Dominic J. Bennett orcid: 0000-0003-2722-1359 affiliation: "1, 2" - name: Hannes Hettling affiliation: "3" - name: Daniele Silvestro orcid: 0000-0003-0100-0961 affiliation: "1, 2" - name: Rutger Vos orcid: 0000-0001-9254-7318 affiliation: "3" - name: Alexandre Antonelli orcid: 0000-0003-1842-9297 affiliation: "1, 2, 4" affiliations: - name: Gothenburg Global Biodiversity Centre, Box 461, SE-405 30 Gothenburg, Sweden index: 1 - name: Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, SE-405 30 Gothenburg, Sweden index: 2 - name: Naturalis Biodiversity Center, P.O. Box 9517, 2300 RA Leiden, The Netherlands index: 3 - name: Gothenburg Botanical Garden, SE 41319 Gothenburg, Sweden index: 4 date: 27 November 2018 bibliography: paper.bib
Summary
Downloading sequences and sequence information from GenBank [@Benson2013] and related NCBI databases is often performed via the NCBI API, Entrez [@Ostell2002]. Entrez, however, has a limit on the number of requests, thus downloading large amounts of sequence data in this way can be inefficient. For situations where a large number of Entrez calls is made, downloading may take days, weeks or even months and could result in a user’s IP address being blacklisted from the NCBI services due to server overload. Additionally, Entrez limits the number of entries that can be retrieved at once, requiring a user to develop code for querying in batches.
The restez package [@restez_z] aims to make sequence retrieval more efficient by allowing a user to download the GenBank database, either in its entirety or in subsets, to their local machine and query this local database instead. This process is more time efficient as GenBank downloads are made via NCBI’s FTP server using compressed sequence files. With a good internet connection and a computer with currently standard capabilities, a database comprising 7 GB of sequence information (i.e. the total sequence data available for Rodentia as of 27 June 2018) can be generated in less than 10 minutes. (For an outline of the functions and structure of restez, see Figure 1.)
Rentrez integration
rentrez [@Winter2017] is a popular R package for querying NCBI’s databases via Entrez in R. To maximize the compatibility of restez, we implemented wrapper functions with the same names and arguments as the rentrez equivalents. Whenever a wrapper function is called the local database copy is searched first. If IDs are missing in the local database a secondary call to Entrez is made via the internet. This allows for easy employment of restez in scripts and packages that are already using rentrez. At a minimum, a user currently using rentrez will only need to create a local subset of the GenBank database, call restez instead of rentrez and ensure the restez database is connected.
Examples
A small example
After a restez database has been set-up, we can retrieve all the sequences from an rentrez::entrez_search() with a single command.
# Use rentrez to search for accession IDs of interest
# Sequences in fasta format can then be retrieved with entrez_fetch
res <- rentrez::entrez_fetch(db = 'nuccore', id = ids, rettype = 'fasta')
# ^ likely to raise an error if too many IDs
res <- restez::entrez_fetch(db = 'nuccore', id = ids, rettype = 'fasta')
# ^ not likely to raise an error
A large example
phylotaR is an R package for retrieving and identifying orthologous sequence clusters from GenBank as a first step in a phylogenetic analysis [@Bennett2018]. Because the package runs an automated pipeline, multiple queries to GenBank via Entrez are made using the rentrez package. As a result, for large taxonomic groups containing well-sequenced organisms the pipeline can take a long time to complete.
library(phylotaR)
# run phylotaR pipeline for New World Monkeys
txid <- 9479 # taxonomic ID
setup(wd = 'nw_monkeys', txid = txid)
run(wd = wd)
# ^ takes around 40 minutes
We can download and create a local copy of the primates GenBank locally and re-run the above code with a library call to restez for speed-up gains and increased code reliability.
# setup database
library(restez)
# Specify path to a local directory in which database will be stored
# Make sure you have sufficient disk space!
restez_path_set(filepath = 'restez_db')
db_download(db = 'nucleotide') # Interactively download GenBank data
db_create(db = 'nucleotide')
Now when re-running the first phylotaR code block with the inclusion of the restez package, the procedure completes approximately eight times faster.
# run phylotaR again
library(phylotaR)
library(restez)
restez_path_set(filepath = 'restez_db')
txid <- 9479
setup(wd = 'nw_monkeys', txid = txid)
run(wd = wd)
# ^ takes around 5 minutes
For more detailed and up-to-date examples and tutorials, see the restez GitHub page [@restez_gh].
Availability
restez is open source software made available under the MIT license. It can be installed through CRAN [@restez_cran], install.package("restez"), or from its GitHub source code repository using the devtools package, e.g. as follows: devtools::install_github("ropensci/restez")
Funding
This package has been developed as part of the supersmartR project [@supersmartR] which has received funding through A.A. (from the Swedish Research Council [B0569601], the Swedish Foundation for Strategic Research, a Wallenberg Academy Fellowship, the Faculty of Sciences at the University of Gothenburg, the Wenner-Gren Foundations, and the David Rockefeller Center for Latin American Studies at Harvard University) and through D.S. (from the Swedish Research Council [2015-04748]).
References
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.