README.md
In robinweide/tagmeppr: A computational pipeline to map tagmap-insertions
tagMeppr 





A computational pipeline to map TagMap-reads
TagMap is a very useful method for transposon mapping (Stern 2017),
enabling researchers to map the insertion sites with ease and generate
long sequencing reads. However, there is little to none automatisation
and downstream analysis software available for these reads. TagMeppr is
an easy to use, memory efficient fastq-to-figure package written in R.

Installation
# Install development version from GitHub
devtools::install_github("robinweide/tagmeppr")
The method
Find insertions
The findInsertions() function will first find all reads that overlap a
TIS, which in the case of PiggyBac will be “TTAA”. Next it will
calculate whether there is a bias towards one side of the TIS using a
binominal test. The bias, denoted as
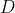 ,
,
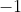 when all reads are
upstream and
when all reads are
upstream and 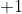 when
all reads are downstream of the TIS.This is done independently for the
forward and reverse reads:
when
all reads are downstream of the TIS.This is done independently for the
forward and reverse reads:

Next, we filter out TISs which have the bias on the same side of the
TIS:

To calculate a “TIS-specific” p-value, we use Edgington’s sum-p method,
which is very conservative in our usage. This ensures that, when
 , both the fwd and the rev reads are indeed
biased.
, both the fwd and the rev reads are indeed
biased.

Afterwards, a holm-correction is done to limit the Family-Wise Error
Rate (FWER).
Usage
The basic usage of tagMapper revolves around three clear steps:
- index: a tagMapper-index is made once for a specific genome and
protocol (e.g. hg19 and PigyBac).
- align: a tagMapperSample-object is made and aligned to the index
- analyse: determine and plot highly likely integraton-sites
Within the analyse-step, you can choose to look at individually found
insertion- sites with plotSite() to check the read-distribution. Here,
reads from the forward and reverse primers overlapping the Target
Insertion Site (TIS) are sorted. This can be helpfull for
quality-checking and determining if the protocol behaves as expected. In
the top-right corner is some important information about the selected
hit: the two D-scores (denoting the bias of up- and downstream mapping
of forward and reverse reads) and the
probability.

You can also look at all found sites in one ideogram with
plotInsertions(), subsetted on the orientation of the insertion and/or
multiple
samples.

See the
vignette
for a more in-depth coverage of all things tagMeppr!
BWA not found
Users with no root-access can install BWA themselves with bioconda or miniconda3. However, Rstudio will have troubles finding BWA. To fix this, run align() and makeIndex with the following:
library(withr)
with_path("/DATA/usr/r.weide/miniconda3/bin",
{align(exp = mysample,
ref = reference_mm10_TM,
cores = 30)})
Code of conduct
Please note that this project is released with a Contributor Code of
Conduct. By participating in this project
you agree to abide by its terms.
Stern, David L. 2017. “Tagmentation-Based Mapping (Tagmap) of Mobile Dna
Genomic Insertion Sites.” *bioRxiv*. Cold Spring Harbor Laboratory.
.
robinweide/tagmeppr documentation built on Feb. 26, 2020, 10:41 p.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
tagMeppr
![]()
A computational pipeline to map TagMap-reads
TagMap is a very useful method for transposon mapping (Stern 2017), enabling researchers to map the insertion sites with ease and generate long sequencing reads. However, there is little to none automatisation and downstream analysis software available for these reads. TagMeppr is an easy to use, memory efficient fastq-to-figure package written in R.
Installation
# Install development version from GitHub
devtools::install_github("robinweide/tagmeppr")
The method
Find insertions
The findInsertions() function will first find all reads that overlap a
TIS, which in the case of PiggyBac will be “TTAA”. Next it will
calculate whether there is a bias towards one side of the TIS using a
binominal test. The bias, denoted as
,
when all reads are
upstream and
when
all reads are downstream of the TIS.This is done independently for the
forward and reverse reads:
Next, we filter out TISs which have the bias on the same side of the TIS:
To calculate a “TIS-specific” p-value, we use Edgington’s sum-p method,
which is very conservative in our usage. This ensures that, when
, both the fwd and the rev reads are indeed
biased.
Afterwards, a holm-correction is done to limit the Family-Wise Error Rate (FWER).
Usage
The basic usage of tagMapper revolves around three clear steps:
- index: a tagMapper-index is made once for a specific genome and protocol (e.g. hg19 and PigyBac).
- align: a tagMapperSample-object is made and aligned to the index
- analyse: determine and plot highly likely integraton-sites
Within the analyse-step, you can choose to look at individually found
insertion- sites with plotSite() to check the read-distribution. Here,
reads from the forward and reverse primers overlapping the Target
Insertion Site (TIS) are sorted. This can be helpfull for
quality-checking and determining if the protocol behaves as expected. In
the top-right corner is some important information about the selected
hit: the two D-scores (denoting the bias of up- and downstream mapping
of forward and reverse reads) and the
probability.
You can also look at all found sites in one ideogram with
plotInsertions(), subsetted on the orientation of the insertion and/or
multiple
samples.
See the vignette for a more in-depth coverage of all things tagMeppr!
BWA not found
Users with no root-access can install BWA themselves with bioconda or miniconda3. However, Rstudio will have troubles finding BWA. To fix this, run align() and makeIndex with the following:
library(withr)
with_path("/DATA/usr/r.weide/miniconda3/bin",
{align(exp = mysample,
ref = reference_mm10_TM,
cores = 30)})
Code of conduct
Please note that this project is released with a Contributor Code of Conduct. By participating in this project you agree to abide by its terms.
Stern, David L. 2017. “Tagmentation-Based Mapping (Tagmap) of Mobile Dna
Genomic Insertion Sites.” *bioRxiv*. Cold Spring Harbor Laboratory.
.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.