README.md
In TearsWillFall/DNAfrags: Filter DNA fragments in BAM files by length and location
DNAfrags
DNAfrags is a group of tools to analyze fragment length in BAM files.
1. Description
DNAfrags simplifies the process of filtering, trimming and visualizing the fragment length, by internally making use of Samtools command-line tricks to access the information within BAM files.
Tools:
htslib: C library for high-throughput sequencing data formats
samtools:Tools (written in C using htslib) for manipulating next-generation sequencing data
libgtextutils: The FASTX-Toolkit text manipulation library
fastx_toolkit: The FASTX-Toolkit is a collection of command line tools for Short-Reads FASTA/FASTQ files preprocessing
2. System Requirements
Tested on fresh Ubuntu and Arch Linux installations. Should be working on other Linux distros too as long as equivalent packages are provided.
In order to be able to download and compile the source files of all the required tools the following programs are required:
git
make
gcc
autoconf
* libtool
These tools can and should be installed using the terminal with the following commands:
- For Ubuntu:
sudo apt install git make gcc autoconf libtool
- For Arch Linux:
sudo pacman -S git make gcc autoconf libtool
Additional dependencies may be needed to succesfully install devtools package in R:
- For Ubuntu:
sudo apt install build-essential libcurl4-gnutls-dev libxml2-dev openssl
- For Arch Linux:
sudo pacman -S build-essential libcurl-gnutls libxml2-dev openssl
3. Installation Instructions
In order to install DNAfrags package we will be using R devtools:
install.packages("devtools")
devtools::install_github("TearsWillFall/DNAfrags")
If devtools package installation fails check System Requirements section, as you may be missing a dependency.
Once the DNAfrags package is installed we can use the function install_required_tools() to download and set up all the tools required for the bioinformatic process. This will create a directory named tools in the current working directory with all the tools. Note: All functions within this package call scripts from the tools directory, therefore if this directory is moved, deleted or the current working directory is changed, these functions will fail.
DNAfrags::install_required_tools()
Or, alternatively.
library("DNAfrags")
install_required_tools()
4. Usage
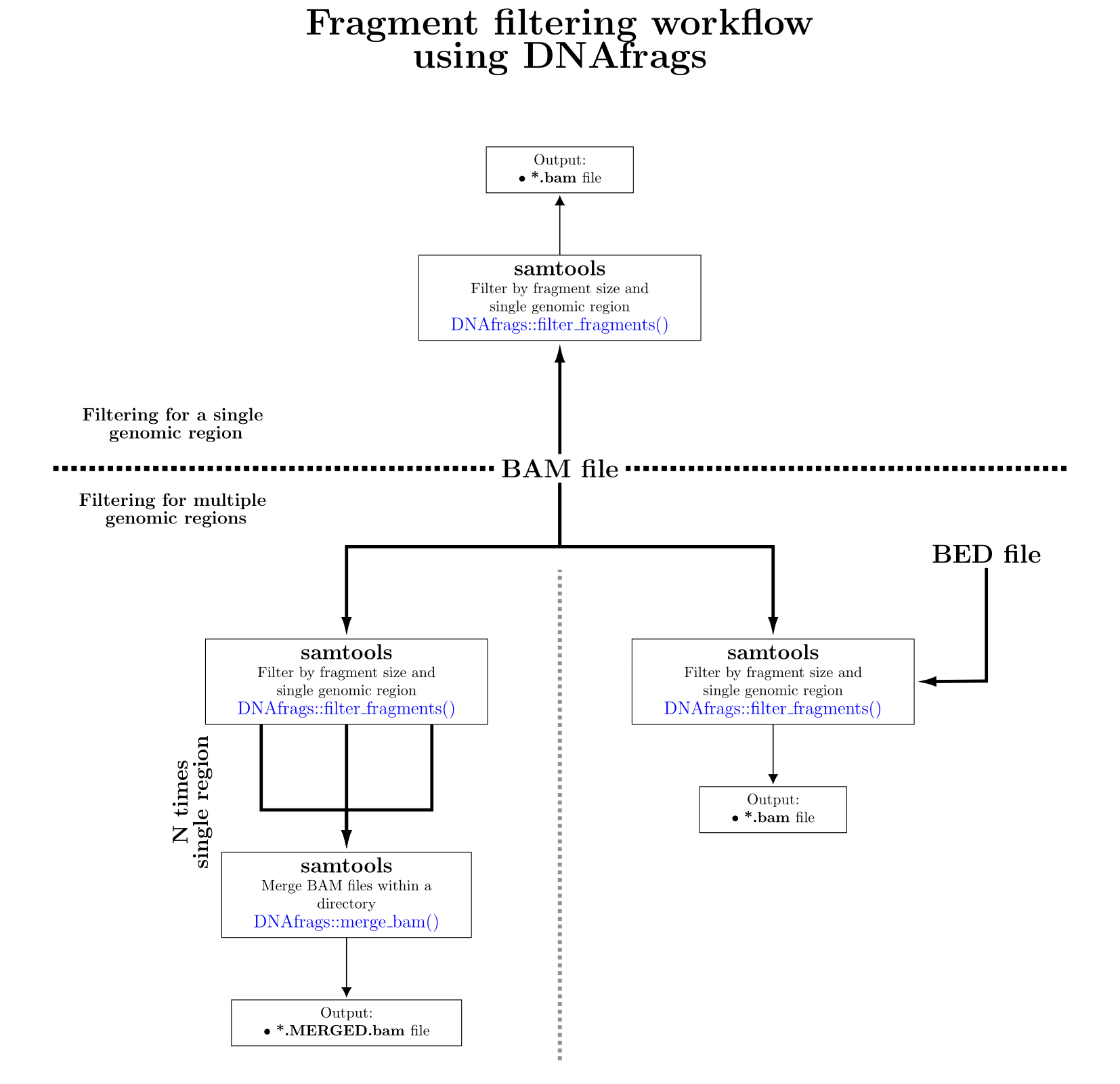
FILTERING:
Examples:
In this example reads in the BAM file called "myBAM.bam" are only filtered by fragment length. Only fragments between lengths 10 and 150 (inclusive) will be kept for all chromosomes.
DNAfrags::filter_fragments(file="myBAM.bam",min_frag_size=10,max_frag_size=150)
In this example reads in the BAM file called "myBAM.bam" are filtered by fragment length and a single genomic regions. Only fragments mapped across chromosome 6 between lengths 10 and 150 (inclusive) will be kept.
DNAfrags::filter_fragments(file="myBAM.bam",min_frag_size=10,max_frag_size=150,chr=6)
In this example reads in the BAM file called "myBAM.bam" are filtered by fragment length and a single genomic regions. Only fragments mapped after the initial 5566 bases in chromosome 6 between lengths 10 and 150 (inclusive) will be kept.
DNAfrags::filter_fragments(file="myBAM.bam",min_frag_size=10,max_frag_size=150,chr=6,start_pos=5566)
In this example reads in the BAM file called "myBAM.bam" are filtered by fragment length and a single genomic regions. Only fragments mapped between the bases 5566 and 1000000 (inclusive) in chromosome 6 between lengths 10 and 150 (inclusive) will be kept.
DNAfrags::filter_fragments(file="myBAM.bam",min_frag_size=10,max_frag_size=150,chr=6,start_pos=5566,end_pos=1000000)
Note:To deal with multiple genomic regions 2 solutions exist
- BED solution [Faster]
In this example reads in the BAM file called "myBAM.bam" are filtered by fragment length in multiple genomic regions. Fragments between lengths 10 and 150 (inclusive) will be kept for specified regions.
DNAfrags::filter_fragments(file="myBAM.bam",min_frag_size=10,max_frag_size=150,bed="myBED.bed")
- Merge BAM solution
In this example reads in the BAM file called "myBAM.bam" are filtered by fragment length in multiple genomic regions. Fragments between lengths 10 and 150 (inclusive) will be kept for specified regions.
DNAfrags::filter_fragments(file="myBAM.bam",min_frag_size=10,max_frag_size=150,chr=6,start_pos=5566)
DNAfrags::filter_fragments(file="myBAM.bam",min_frag_size=10,max_frag_size=150,chr=12,start_pos=123123,end_pos=1232131)
Then the BAM files for each genomic region are merged together into a single BAM file.
DNAfrags::merge_bam(out_bam="MyBAM",bam_dir=".")
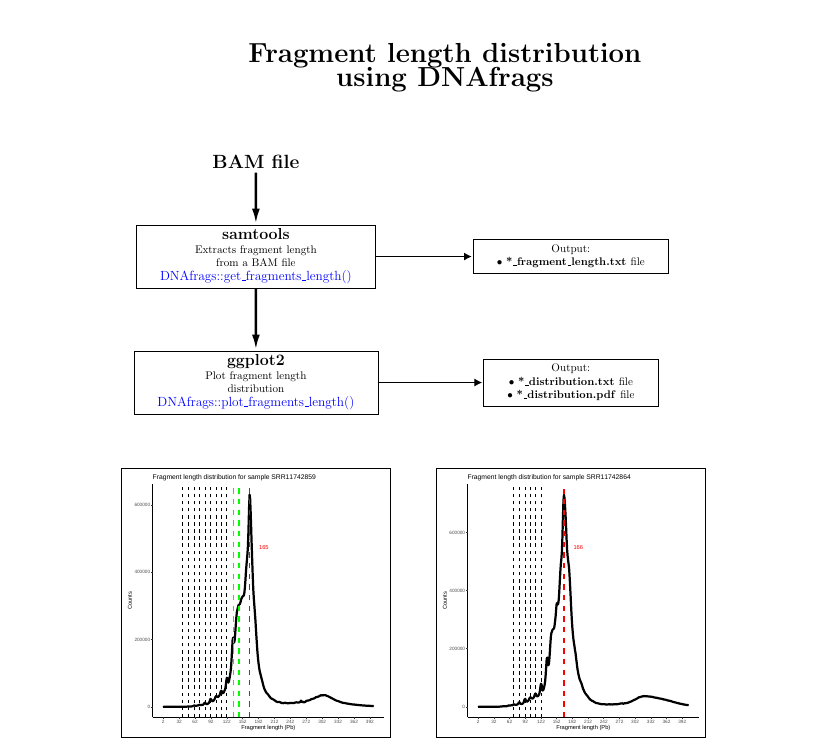
VISUALIZATION:
Examples:
In this example, the fragment length from all the reads in "myBAM.bam" are extracted and saved as *_fragment_length.txt
DNAfrags::get_fragments_length(file="myBAM.bam")
Once the fragment length is extracted, it can be plotted using the plot_fragments_length, as seen below.
DNAfrags::plot_fragments_length(file="mybam_fragment_length.txt")
TearsWillFall/DNAfrags documentation built on March 26, 2022, 6:02 a.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
DNAfrags
DNAfrags is a group of tools to analyze fragment length in BAM files.
1. Description
DNAfrags simplifies the process of filtering, trimming and visualizing the fragment length, by internally making use of Samtools command-line tricks to access the information within BAM files.
Tools: htslib: C library for high-throughput sequencing data formats samtools:Tools (written in C using htslib) for manipulating next-generation sequencing data libgtextutils: The FASTX-Toolkit text manipulation library fastx_toolkit: The FASTX-Toolkit is a collection of command line tools for Short-Reads FASTA/FASTQ files preprocessing
2. System Requirements
Tested on fresh Ubuntu and Arch Linux installations. Should be working on other Linux distros too as long as equivalent packages are provided.
In order to be able to download and compile the source files of all the required tools the following programs are required: git make gcc autoconf * libtool
These tools can and should be installed using the terminal with the following commands:
- For Ubuntu:
sudo apt install git make gcc autoconf libtool
- For Arch Linux:
sudo pacman -S git make gcc autoconf libtool
Additional dependencies may be needed to succesfully install devtools package in R:
- For Ubuntu:
sudo apt install build-essential libcurl4-gnutls-dev libxml2-dev openssl
- For Arch Linux:
sudo pacman -S build-essential libcurl-gnutls libxml2-dev openssl
3. Installation Instructions
In order to install DNAfrags package we will be using R devtools:
install.packages("devtools")
devtools::install_github("TearsWillFall/DNAfrags")
If devtools package installation fails check System Requirements section, as you may be missing a dependency.
Once the DNAfrags package is installed we can use the function install_required_tools() to download and set up all the tools required for the bioinformatic process. This will create a directory named tools in the current working directory with all the tools. Note: All functions within this package call scripts from the tools directory, therefore if this directory is moved, deleted or the current working directory is changed, these functions will fail.
DNAfrags::install_required_tools()
Or, alternatively.
library("DNAfrags")
install_required_tools()
4. Usage
FILTERING:
Examples:
In this example reads in the BAM file called "myBAM.bam" are only filtered by fragment length. Only fragments between lengths 10 and 150 (inclusive) will be kept for all chromosomes.
DNAfrags::filter_fragments(file="myBAM.bam",min_frag_size=10,max_frag_size=150)
In this example reads in the BAM file called "myBAM.bam" are filtered by fragment length and a single genomic regions. Only fragments mapped across chromosome 6 between lengths 10 and 150 (inclusive) will be kept.
DNAfrags::filter_fragments(file="myBAM.bam",min_frag_size=10,max_frag_size=150,chr=6)
In this example reads in the BAM file called "myBAM.bam" are filtered by fragment length and a single genomic regions. Only fragments mapped after the initial 5566 bases in chromosome 6 between lengths 10 and 150 (inclusive) will be kept.
DNAfrags::filter_fragments(file="myBAM.bam",min_frag_size=10,max_frag_size=150,chr=6,start_pos=5566)
In this example reads in the BAM file called "myBAM.bam" are filtered by fragment length and a single genomic regions. Only fragments mapped between the bases 5566 and 1000000 (inclusive) in chromosome 6 between lengths 10 and 150 (inclusive) will be kept.
DNAfrags::filter_fragments(file="myBAM.bam",min_frag_size=10,max_frag_size=150,chr=6,start_pos=5566,end_pos=1000000)
Note:To deal with multiple genomic regions 2 solutions exist
- BED solution [Faster]
In this example reads in the BAM file called "myBAM.bam" are filtered by fragment length in multiple genomic regions. Fragments between lengths 10 and 150 (inclusive) will be kept for specified regions.
DNAfrags::filter_fragments(file="myBAM.bam",min_frag_size=10,max_frag_size=150,bed="myBED.bed")
- Merge BAM solution
In this example reads in the BAM file called "myBAM.bam" are filtered by fragment length in multiple genomic regions. Fragments between lengths 10 and 150 (inclusive) will be kept for specified regions.
DNAfrags::filter_fragments(file="myBAM.bam",min_frag_size=10,max_frag_size=150,chr=6,start_pos=5566)
DNAfrags::filter_fragments(file="myBAM.bam",min_frag_size=10,max_frag_size=150,chr=12,start_pos=123123,end_pos=1232131)
Then the BAM files for each genomic region are merged together into a single BAM file.
DNAfrags::merge_bam(out_bam="MyBAM",bam_dir=".")
VISUALIZATION:
Examples:
In this example, the fragment length from all the reads in "myBAM.bam" are extracted and saved as *_fragment_length.txt
DNAfrags::get_fragments_length(file="myBAM.bam")
Once the fragment length is extracted, it can be plotted using the plot_fragments_length, as seen below.
DNAfrags::plot_fragments_length(file="mybam_fragment_length.txt")
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.