inst/examples/labrid_data.md
In cboettig/wrightscape: wrightscape
labrid data processing
require(phytools)
require(geiger)
require(wrightscape)
(This data has not been released)
labrid_tree <- read.nexus("../../data/labrid_tree.nex")
#fin_data <-read.csv(paste(path,"labrid.csv", sep="")) # not actually being used
diet_data <- read.csv("../../data/labriddata_parrotfish.csv")
Simple size corrections length and weight as fraction of body mass could use
for(i in c(3,4,6,7,8)){
diet_data[i] <- diet_data[i]/diet_data[5]
}
diet_data[5] <- log(diet_data[5])
We will take a different approach, using the phylogenetic correction, so the above code is not to be run.
corrected_data <- diet_data
Use the simple trait names from @Price2010
traitnames <- c("Species", "group", "gape", "prot", "bodymass", "AM", "SH", "LP", "close", "open", "kt")
names(corrected_data) <- traitnames
Lengths are log transformed
corrected_data[["gape"]] <- log(corrected_data[["gape"]])
corrected_data[["prot"]] <- log(corrected_data[["prot"]])
masses are log(cube-root) transformed
corrected_data[["bodymass"]] <- log(corrected_data[["bodymass"]])/3
corrected_data[["AM"]] <- log(corrected_data[["AM"]])/3
corrected_data[["SH"]] <- log(corrected_data[["SH"]])/3
corrected_data[["LP"]] <- log(corrected_data[["LP"]])/3
Traits which are ratios are fine as they are
Drop any unmatched tip-traits
ape <- treedata(labrid_tree, corrected_data[,3:11], corrected_data[,1])
Dropped rows from the data because there were no matching tips in the tree:
[1] Calotomus_carolinus Coris_aurilineata
[3] Halichoeres_melasmapomus Leptoscarus_vaigiensis
[5] Scarus_forsteni Scarus_quoyi
[7] Scarus_rubroviolaceus Xiphocheilus_typus
122 Levels: Anampses_caeruleopunctatus ... Xyrichtys_novacula
Run Revell's phylogenetic size corrections
ape$data["bodysize"]
[1] NA
out <- phyl.resid(ape$phy, ape$data[,"bodymass"], ape$data[,c("gape", "prot","AM", "SH", "LP")] )
The phyl.resid function changes order of species listing. Merge for a set of uncorrected and corrected traits.
traits <- merge(ape$data, out$resid, by="row.names")
columns that are transformed now have gape.x for untransformed, gape.y for transformed.
format_data() gets regimes from column specified in
"regimes" (e.g. this is a column id, not a # of regimes)
This also converts the tree and data into ouch format
(We could just hand it all traits, but these are just the tranformed and size-corrected ones)
labrid <- format_data(labrid_tree, traits[,2:length(traits)], species_names=traits[,1])
Painting Regimes
Select common ancestor of a Chlorurus and a Hipposcarus as the changepoint
intra_ancestor <- mrcaOUCH(c("Chlorurus_sordidus", "Hipposcarus_longiceps"), labrid$tree)
intramandibular <- paintBranches(intra_ancestor, labrid$tree, c("other","intramandibular"))
Select common ancestor for all parrot fish:
pharyngeal_ancestor <- mrcaOUCH(c("Bolbometopon_muricatum", "Sparisoma_radians"), labrid$tree)
pharyngeal <- paintBranches(pharyngeal_ancestor, labrid$tree, c("other","pharyngeal"))
two_shifts <- paintBranches(c(pharyngeal_ancestor, intra_ancestor), labrid$tree, c("wrasses", "pharyngeal", "intramandibular") )
This leaves the branch on which the second transition occurs unspecified (fourth regime).
We have to fix this manually
two_shifts[as.numeric(intra_ancestor)] <- "intramandibular"
two_shifts <- as.factor(as.character(two_shifts))
names(two_shifts) <- names(intramandibular)
rename the ouch-formated data
dat <- labrid$data
tree <- labrid$tree
rename the ape-formatted data
ape.phy <- ape$phy
ape.dat <- traits
Save the final output. (This data is provided with the package).
save(list=c("intramandibular", "pharyngeal", "two_shifts", "tree", "dat", "ape.phy", "ape.dat"), file="labrids.rda")
We now have access to the following configurations:
intramandibular, pharyngeal and two_shifts paintings, (and labrid$noregimes),
and tree in labrid$tree
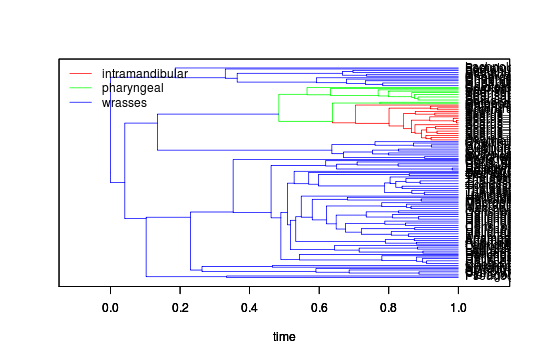
plot(labrid$tree, regimes=two_shifts)
cboettig/wrightscape documentation built on May 13, 2019, 2:12 p.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
labrid data processing
require(phytools)
require(geiger)
require(wrightscape)
(This data has not been released)
labrid_tree <- read.nexus("../../data/labrid_tree.nex")
#fin_data <-read.csv(paste(path,"labrid.csv", sep="")) # not actually being used
diet_data <- read.csv("../../data/labriddata_parrotfish.csv")
Simple size corrections length and weight as fraction of body mass could use
for(i in c(3,4,6,7,8)){
diet_data[i] <- diet_data[i]/diet_data[5]
}
diet_data[5] <- log(diet_data[5])
We will take a different approach, using the phylogenetic correction, so the above code is not to be run.
corrected_data <- diet_data
Use the simple trait names from @Price2010
traitnames <- c("Species", "group", "gape", "prot", "bodymass", "AM", "SH", "LP", "close", "open", "kt")
names(corrected_data) <- traitnames
Lengths are log transformed
corrected_data[["gape"]] <- log(corrected_data[["gape"]])
corrected_data[["prot"]] <- log(corrected_data[["prot"]])
masses are log(cube-root) transformed
corrected_data[["bodymass"]] <- log(corrected_data[["bodymass"]])/3
corrected_data[["AM"]] <- log(corrected_data[["AM"]])/3
corrected_data[["SH"]] <- log(corrected_data[["SH"]])/3
corrected_data[["LP"]] <- log(corrected_data[["LP"]])/3
Traits which are ratios are fine as they are
Drop any unmatched tip-traits
ape <- treedata(labrid_tree, corrected_data[,3:11], corrected_data[,1])
Dropped rows from the data because there were no matching tips in the tree:
[1] Calotomus_carolinus Coris_aurilineata
[3] Halichoeres_melasmapomus Leptoscarus_vaigiensis
[5] Scarus_forsteni Scarus_quoyi
[7] Scarus_rubroviolaceus Xiphocheilus_typus
122 Levels: Anampses_caeruleopunctatus ... Xyrichtys_novacula
Run Revell's phylogenetic size corrections
ape$data["bodysize"]
[1] NA
out <- phyl.resid(ape$phy, ape$data[,"bodymass"], ape$data[,c("gape", "prot","AM", "SH", "LP")] )
The phyl.resid function changes order of species listing. Merge for a set of uncorrected and corrected traits.
traits <- merge(ape$data, out$resid, by="row.names")
columns that are transformed now have gape.x for untransformed, gape.y for transformed.
format_data() gets regimes from column specified in
"regimes" (e.g. this is a column id, not a # of regimes)
This also converts the tree and data into ouch format
(We could just hand it all traits, but these are just the tranformed and size-corrected ones)
labrid <- format_data(labrid_tree, traits[,2:length(traits)], species_names=traits[,1])
Painting Regimes
Select common ancestor of a Chlorurus and a Hipposcarus as the changepoint
intra_ancestor <- mrcaOUCH(c("Chlorurus_sordidus", "Hipposcarus_longiceps"), labrid$tree)
intramandibular <- paintBranches(intra_ancestor, labrid$tree, c("other","intramandibular"))
Select common ancestor for all parrot fish:
pharyngeal_ancestor <- mrcaOUCH(c("Bolbometopon_muricatum", "Sparisoma_radians"), labrid$tree)
pharyngeal <- paintBranches(pharyngeal_ancestor, labrid$tree, c("other","pharyngeal"))
two_shifts <- paintBranches(c(pharyngeal_ancestor, intra_ancestor), labrid$tree, c("wrasses", "pharyngeal", "intramandibular") )
This leaves the branch on which the second transition occurs unspecified (fourth regime). We have to fix this manually
two_shifts[as.numeric(intra_ancestor)] <- "intramandibular"
two_shifts <- as.factor(as.character(two_shifts))
names(two_shifts) <- names(intramandibular)
rename the ouch-formated data
dat <- labrid$data
tree <- labrid$tree
rename the ape-formatted data
ape.phy <- ape$phy
ape.dat <- traits
Save the final output. (This data is provided with the package).
save(list=c("intramandibular", "pharyngeal", "two_shifts", "tree", "dat", "ape.phy", "ape.dat"), file="labrids.rda")
We now have access to the following configurations:
intramandibular, pharyngeal and two_shifts paintings, (and labrid$noregimes),
and tree in labrid$tree
plot(labrid$tree, regimes=two_shifts)
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.