README.md
In ruqianl/comapr: Crossover analysis and genetic map construction
comapr


Install
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("comapr")
Introduction

comapr is an R package for finding crossovers for SNP marker intervals
by detecting haplotype shifts across groups of samples.
From marker genotyping results in data.frame
comapr can be applied for detecting crossovers from genotyping results
of makers across samples in a marker by sample data.frame.
library(comapr)
head(snp_geno)
#> CHR POS rsID C57BL.6J FVB.NJ..i. X92 X93 X94 X95 X96 X97 X98
#> 1 1 4526088 rs13475701 GG CC CC CC CC CC CC CC CG
#> 2 1 5595513 rs13475705 TT CC CC CC CC CC CC CC CT
#> 3 1 6057774 rs3710263 CC TT TT TT TT TT TT TT TC
#> 4 1 6655964 rs13475709 CC GG GG GG GG GG GG GG CG
#> 5 1 21638464 rs6253968 TT CC Fail CC CC CC CC CC Fail
#> 6 1 22665060 rs6361963 AA GG AG GG GG GG GG GG AG
#> X99 X100 X101 X102 X103 X104 X105 X106 X107 X108 X109 X110 X111 X112 X113
#> 1 CG CC CG CG CC CG CG CC CC CC CC CG CC CC CC
#> 2 CT CC CT CT CC CT CT CC CC CC CC CT CC CC CC
#> 3 TC TT TC TC TT TC TC TT TT TT TT TC TT TT TT
#> 4 CG GG CG CG GG CG CG GG GG GG GG CG GG GG GG
#> 5 Fail CC TC TC CC CC CC CC CC CC Fail TC CC CC CC
#> 6 AG GG AG AG GG GG GG AG GG GG GG AG GG GG GG
From sgcocaller’s sparse matrices
comapr is also engineered to analyze the outputs from a single-sperm
crossover calling tool
sgcocaller
list.files("inst/extdata/")
#> [1] "s1_barcodes.txt" "s1_chr1_altCount.mtx" "s1_chr1_snpAnnot.txt"
#> [4] "s1_chr1_totalCount.mtx" "s1_chr1_vi.mtx" "s1_chr1_viSegInfo.txt"
#> [7] "s1_chr2_altCount.mtx" "s1_chr2_snpAnnot.txt" "s1_chr2_totalCount.mtx"
#> [10] "s1_chr2_vi.mtx" "s1_chr2_viSegInfo.txt" "s1_chr3_altCount.mtx"
#> [13] "s1_chr3_snpAnnot.txt" "s1_chr3_totalCount.mtx" "s1_chr3_vi.mtx"
#> [16] "s1_chr3_viSegInfo.txt" "s1_chr4_altCount.mtx" "s1_chr4_snpAnnot.txt"
#> [19] "s1_chr4_totalCount.mtx" "s1_chr4_vi.mtx" "s1_chr4_viSegInfo.txt"
#> [22] "s1_chr5_altCount.mtx" "s1_chr5_snpAnnot.txt" "s1_chr5_totalCount.mtx"
#> [25] "s1_chr5_vi.mtx" "s1_chr5_viSegInfo.txt" "s2_barcodes.txt"
#> [28] "s2_chr1_snpAnnot.txt" "s2_chr1_vi.mtx" "s2_chr1_viSegInfo.txt"
#> [31] "s2_chr2_snpAnnot.txt" "s2_chr2_vi.mtx" "s2_chr2_viSegInfo.txt"
#> [34] "s2_chr3_snpAnnot.txt" "s2_chr3_vi.mtx" "s2_chr3_viSegInfo.txt"
#> [37] "s2_chr4_snpAnnot.txt" "s2_chr4_vi.mtx" "s2_chr4_viSegInfo.txt"
#> [40] "s2_chr5_snpAnnot.txt" "s2_chr5_vi.mtx" "s2_chr5_viSegInfo.txt"
Visualizing feature of single sperms
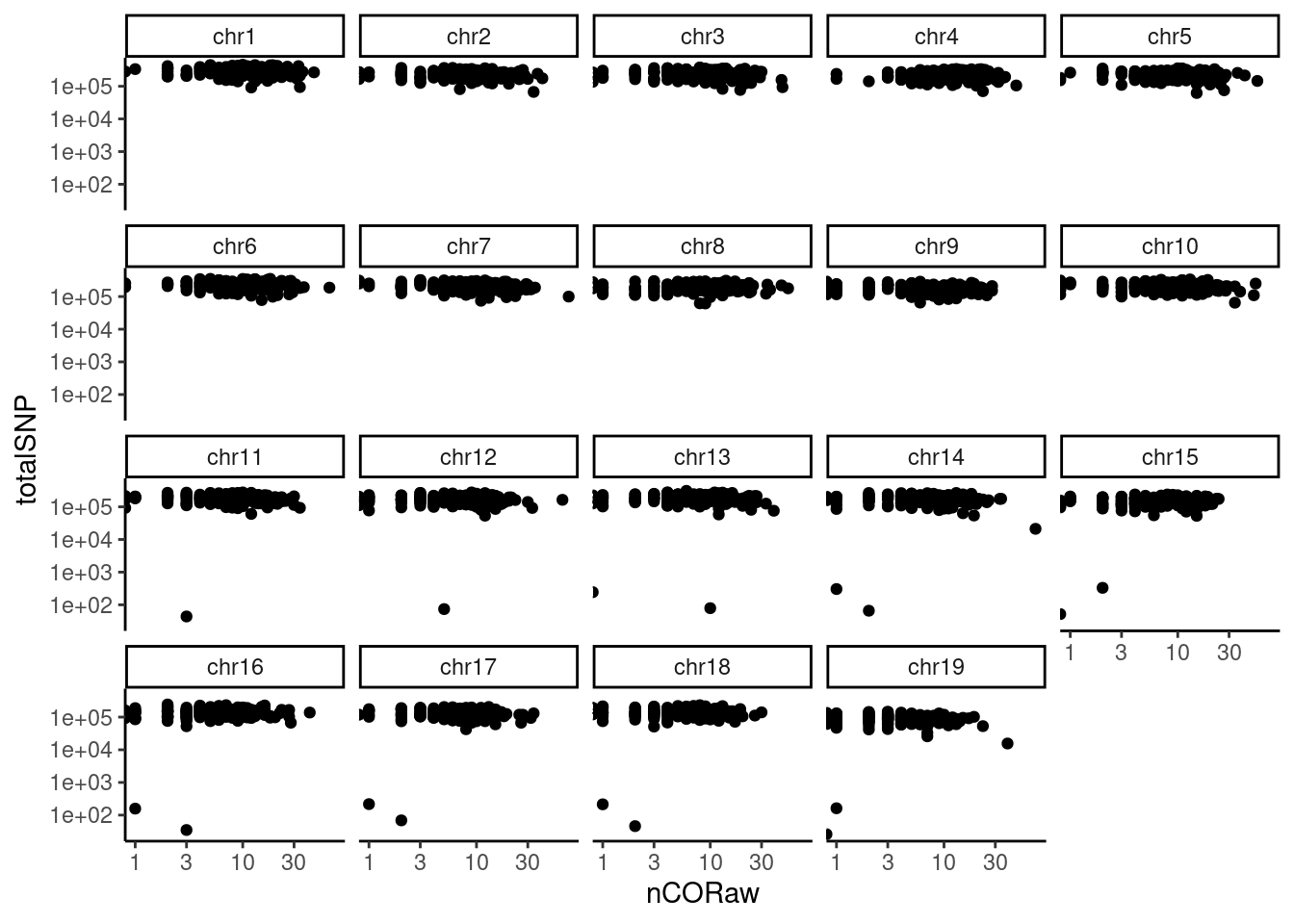
Crossover positions

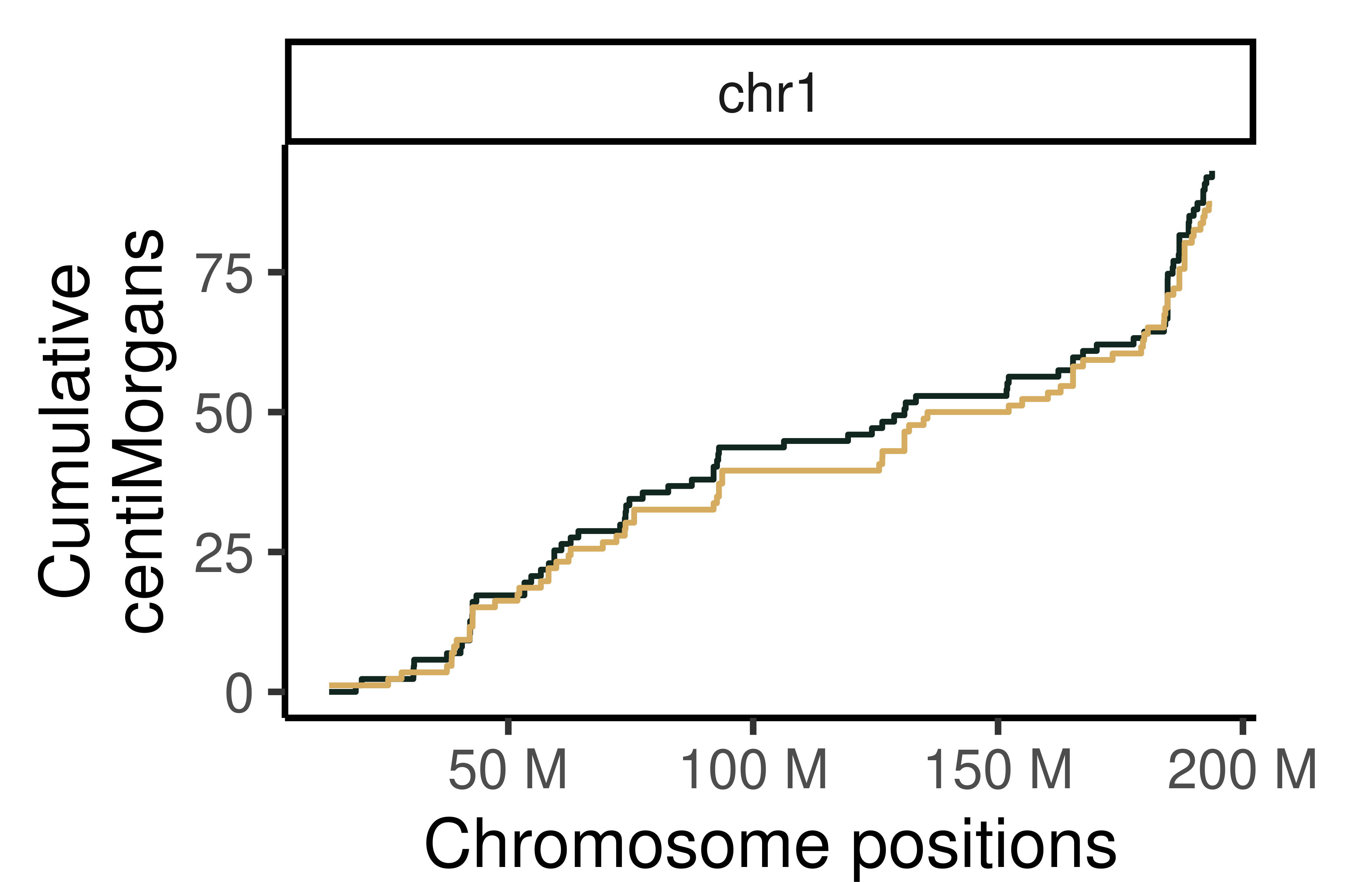
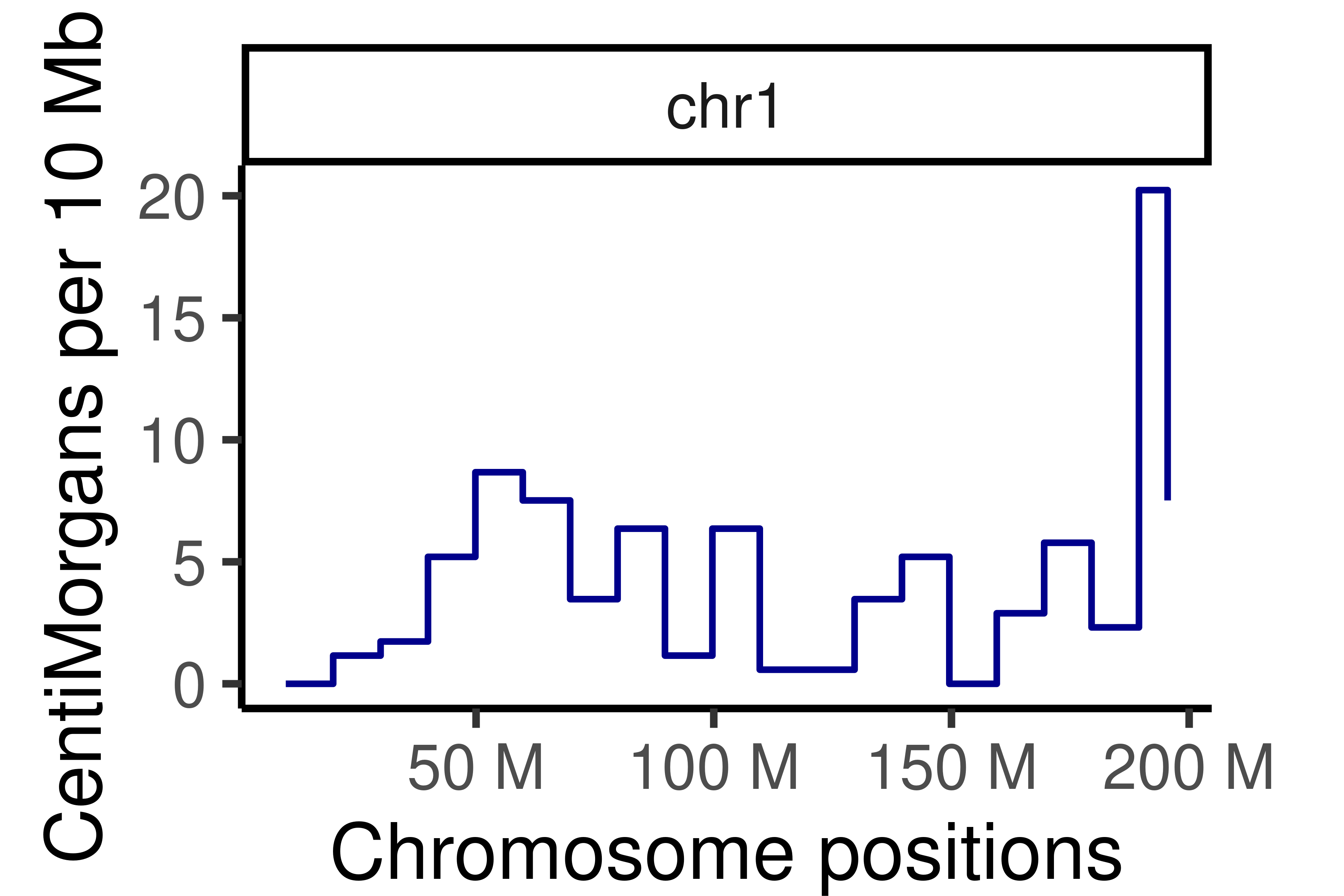
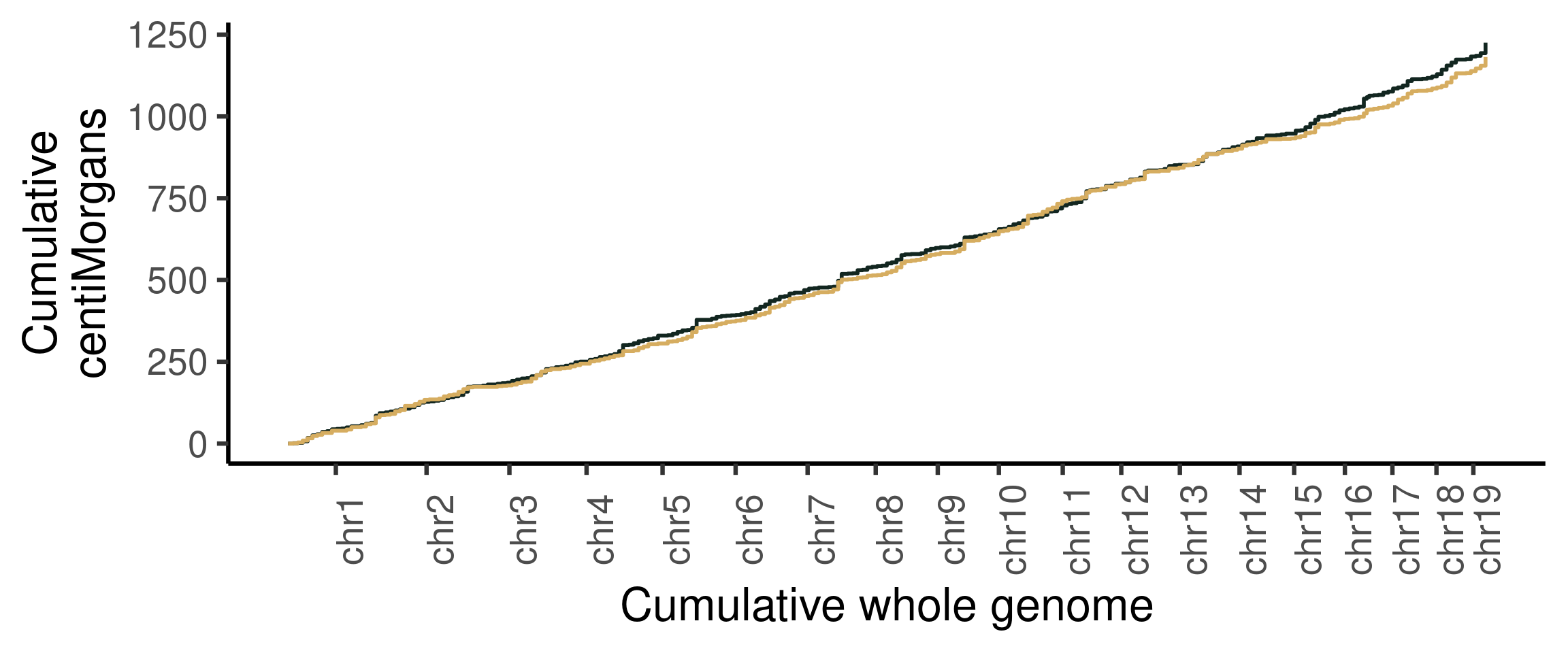
Cumumative Genetic distances across intervals
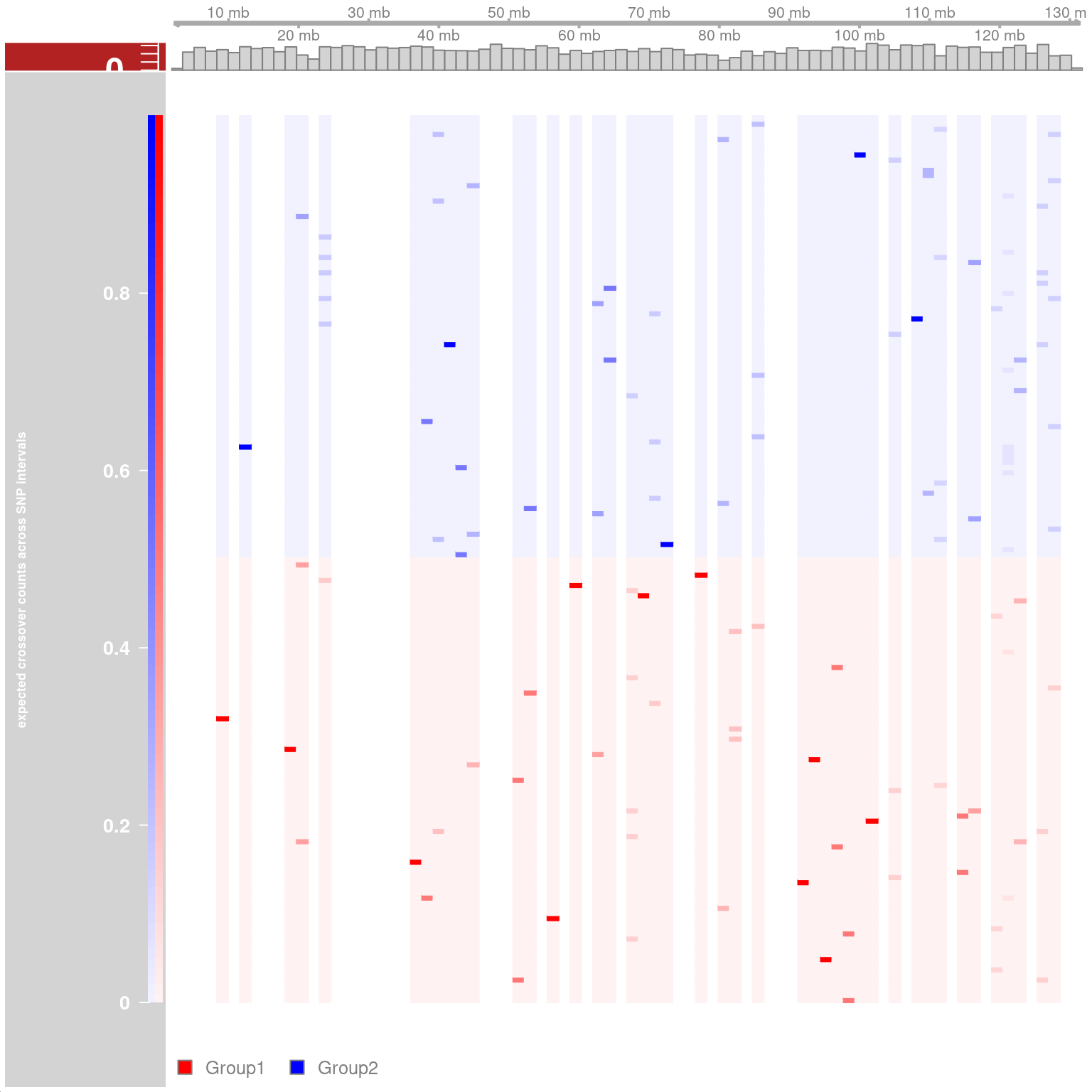
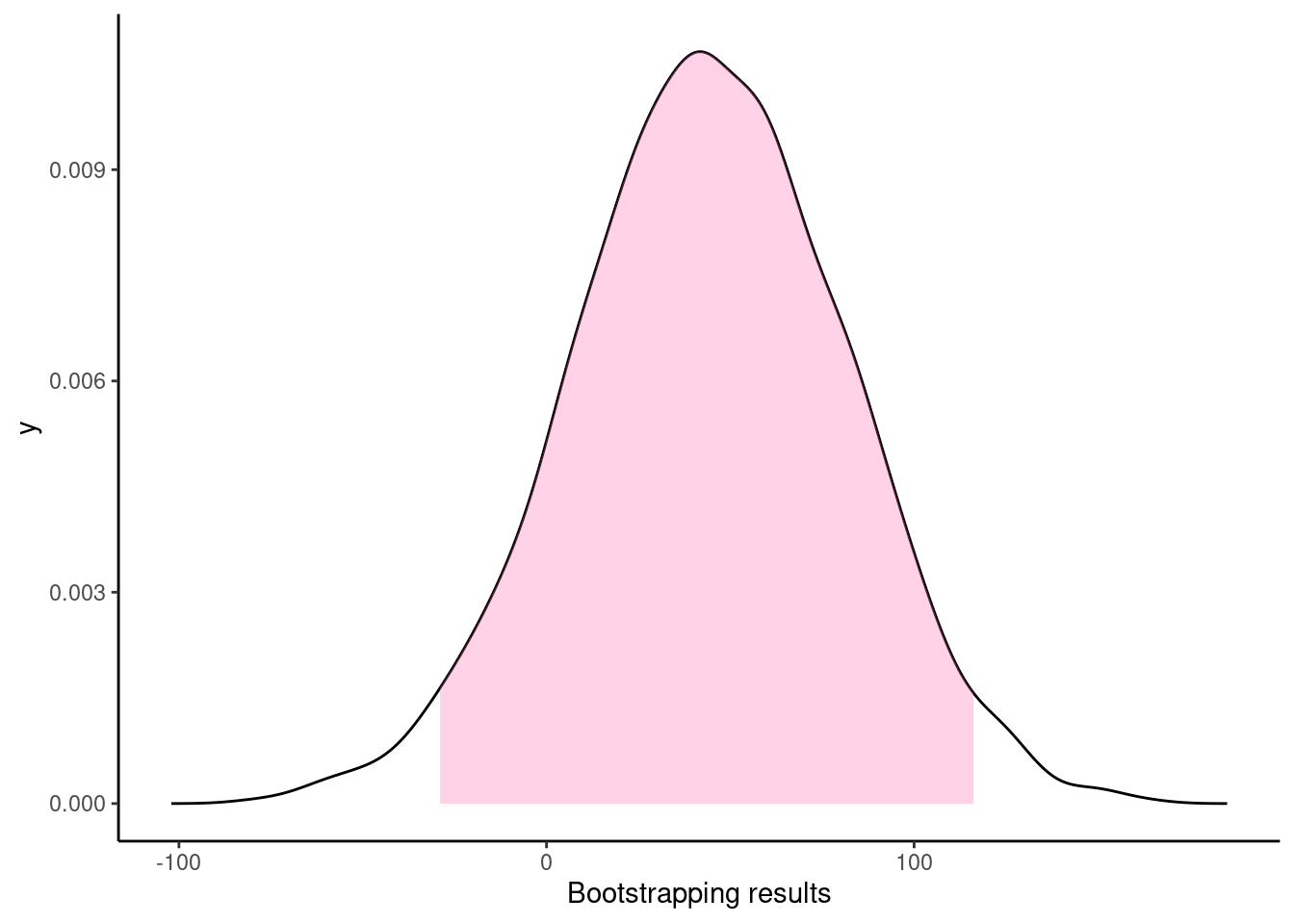
Resampling methods for comparing groups
Analysis workflow for demonstration of comapr on a single-sperm DNAseq dataset
We demonstrate the usage of sgcocaller and comapr for identifying
and visualising crossovers regions from single-sperm DNA sequencing
dataset
here
ruqianl/comapr documentation built on Oct. 27, 2023, 5:12 a.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
comapr
![]()
Install
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("comapr")
Introduction
comapr is an R package for finding crossovers for SNP marker intervals
by detecting haplotype shifts across groups of samples.
From marker genotyping results in data.frame
comapr can be applied for detecting crossovers from genotyping results
of makers across samples in a marker by sample data.frame.
library(comapr)
head(snp_geno)
#> CHR POS rsID C57BL.6J FVB.NJ..i. X92 X93 X94 X95 X96 X97 X98
#> 1 1 4526088 rs13475701 GG CC CC CC CC CC CC CC CG
#> 2 1 5595513 rs13475705 TT CC CC CC CC CC CC CC CT
#> 3 1 6057774 rs3710263 CC TT TT TT TT TT TT TT TC
#> 4 1 6655964 rs13475709 CC GG GG GG GG GG GG GG CG
#> 5 1 21638464 rs6253968 TT CC Fail CC CC CC CC CC Fail
#> 6 1 22665060 rs6361963 AA GG AG GG GG GG GG GG AG
#> X99 X100 X101 X102 X103 X104 X105 X106 X107 X108 X109 X110 X111 X112 X113
#> 1 CG CC CG CG CC CG CG CC CC CC CC CG CC CC CC
#> 2 CT CC CT CT CC CT CT CC CC CC CC CT CC CC CC
#> 3 TC TT TC TC TT TC TC TT TT TT TT TC TT TT TT
#> 4 CG GG CG CG GG CG CG GG GG GG GG CG GG GG GG
#> 5 Fail CC TC TC CC CC CC CC CC CC Fail TC CC CC CC
#> 6 AG GG AG AG GG GG GG AG GG GG GG AG GG GG GG
From sgcocaller’s sparse matrices
comapr is also engineered to analyze the outputs from a single-sperm
crossover calling tool
sgcocaller
list.files("inst/extdata/")
#> [1] "s1_barcodes.txt" "s1_chr1_altCount.mtx" "s1_chr1_snpAnnot.txt"
#> [4] "s1_chr1_totalCount.mtx" "s1_chr1_vi.mtx" "s1_chr1_viSegInfo.txt"
#> [7] "s1_chr2_altCount.mtx" "s1_chr2_snpAnnot.txt" "s1_chr2_totalCount.mtx"
#> [10] "s1_chr2_vi.mtx" "s1_chr2_viSegInfo.txt" "s1_chr3_altCount.mtx"
#> [13] "s1_chr3_snpAnnot.txt" "s1_chr3_totalCount.mtx" "s1_chr3_vi.mtx"
#> [16] "s1_chr3_viSegInfo.txt" "s1_chr4_altCount.mtx" "s1_chr4_snpAnnot.txt"
#> [19] "s1_chr4_totalCount.mtx" "s1_chr4_vi.mtx" "s1_chr4_viSegInfo.txt"
#> [22] "s1_chr5_altCount.mtx" "s1_chr5_snpAnnot.txt" "s1_chr5_totalCount.mtx"
#> [25] "s1_chr5_vi.mtx" "s1_chr5_viSegInfo.txt" "s2_barcodes.txt"
#> [28] "s2_chr1_snpAnnot.txt" "s2_chr1_vi.mtx" "s2_chr1_viSegInfo.txt"
#> [31] "s2_chr2_snpAnnot.txt" "s2_chr2_vi.mtx" "s2_chr2_viSegInfo.txt"
#> [34] "s2_chr3_snpAnnot.txt" "s2_chr3_vi.mtx" "s2_chr3_viSegInfo.txt"
#> [37] "s2_chr4_snpAnnot.txt" "s2_chr4_vi.mtx" "s2_chr4_viSegInfo.txt"
#> [40] "s2_chr5_snpAnnot.txt" "s2_chr5_vi.mtx" "s2_chr5_viSegInfo.txt"
Visualizing feature of single sperms
Crossover positions
Cumumative Genetic distances across intervals
Resampling methods for comparing groups
Analysis workflow for demonstration of comapr on a single-sperm DNAseq dataset
We demonstrate the usage of sgcocaller and comapr for identifying
and visualising crossovers regions from single-sperm DNA sequencing
dataset
here
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.