inst/anvil/DESCRIPTION.md
In vjcitn/oc2bioc: simple interfaces from OpenCRAVAT to Bioconductor
Use rstudio and the oc2bioc R package at github.com/vjcitn to illustrate variant annotation
Setting up the environment
We are using the default community-maintained rstudio environment as of 10 Jan 2021
In a terminal, run the commands
pip3 install open-cravat
.local/bin/oc module install-base
.local/bin/oc module install segway_lung clinvar
Then in the Rstudio console, issue the command
BiocManager::install("vjcitn/oc2bioc")
For illustrations given below we will need two more packages:
ii = rownames(installed.packages())
if (!("curatedTCGAData"%in% ii)) BiocManager::install("curatedTCGAData")
if (!("RaggedExperiment" %in% ii)) BiocManager::install("RaggedExperiment")
Acquiring mutation data for adrenocortical carcinoma from TCGA
The curatedTCGAData package provides easy access to mutations collected on 36 tumor types.
We'll collect mutations on the ACC tumors:
library(curatedTCGAData) # install if necessary
library(RaggedExperiment)
suppressMessages({
acc = curatedTCGAData("ACC", "Mutation", dry.run=FALSE, verbose=FALSE)
})
mut = experiments(acc)[[1]]
mut
The "assays" available record much information about the
specific variants; we only need REF/ALT codes.
```{r theass,eval=FALSE}
assayNames(mut)[c(1,7:9)]
The addresses of the variants are available after coercing
the `RaggedArray` instance to a `GRangesList`; this is simplified
to a `GRanges` with `unlist`.
```{r geta,eval=FALSE}
gr = unlist(as(mut, "GRangesList"))
dim(mcols(gr))
head(gr[,1:3],3)
Multibase variants are present. We'll
confine attention to single-nucleotide variants (SNV).
```{r getsnv, eval=FALSE}
table(width(gr))[1:6]
gr = gr[width(gr)==1]
We'll want the variants in GRCh38 coordinates.
```{r doma,eval=FALSE}
seqlevels(gr) = paste0("chr", seqlevels(gr))
gr38 = unlist(rtracklayer::liftOver(gr, oc2bioc::ch19to38))
genome(gr38) = "hg38" # UCSC terminology
head(gr38[,1:3],3)
length(gr38)
length(unique(gr38))
Transforming variants to a TSV file for use with R and OpenCRAVAT
```{r mm}
library(oc2bioc)
mtb = make_oc_POSTable(gr38) # already remapped
head(mtb)
Use code like
write.table(mtb, file="/home/rstudio/accdemo.tsv", sep="\t", col.names=FALSE, row.names=FALSE, quote=FALSE)
to place the formatted mutation data where it is easily found.
Now, invoke the shiny app for OpenCRAVAT annotation and reporting:
oc2bioc(".local/bin/cravat")
```
Click on the radio button corresponding to the tsv file created above. The following will eventually appear:

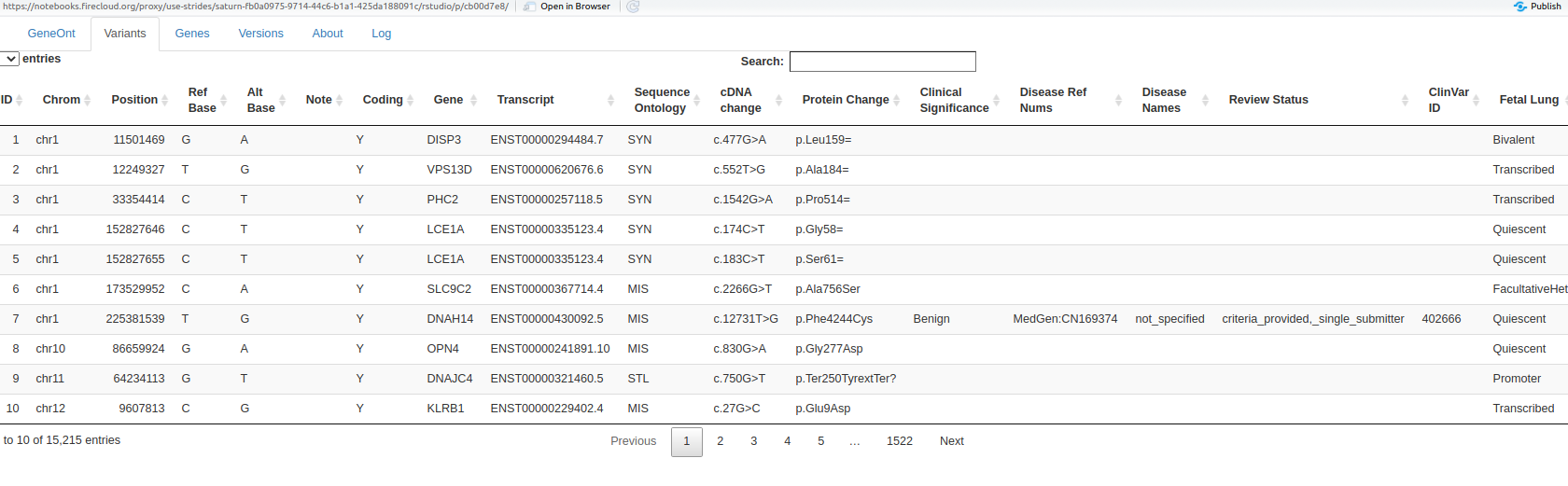
Details on the unique variants reported are found at the variants tab:
vjcitn/oc2bioc documentation built on Sept. 7, 2024, 10:23 a.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Use rstudio and the oc2bioc R package at github.com/vjcitn to illustrate variant annotation
Setting up the environment
We are using the default community-maintained rstudio environment as of 10 Jan 2021
In a terminal, run the commands
pip3 install open-cravat
.local/bin/oc module install-base
.local/bin/oc module install segway_lung clinvar
Then in the Rstudio console, issue the command
BiocManager::install("vjcitn/oc2bioc")
For illustrations given below we will need two more packages:
ii = rownames(installed.packages())
if (!("curatedTCGAData"%in% ii)) BiocManager::install("curatedTCGAData")
if (!("RaggedExperiment" %in% ii)) BiocManager::install("RaggedExperiment")
Acquiring mutation data for adrenocortical carcinoma from TCGA
The curatedTCGAData package provides easy access to mutations collected on 36 tumor types. We'll collect mutations on the ACC tumors:
library(curatedTCGAData) # install if necessary
library(RaggedExperiment)
suppressMessages({
acc = curatedTCGAData("ACC", "Mutation", dry.run=FALSE, verbose=FALSE)
})
mut = experiments(acc)[[1]]
mut
The "assays" available record much information about the specific variants; we only need REF/ALT codes. ```{r theass,eval=FALSE} assayNames(mut)[c(1,7:9)]
The addresses of the variants are available after coercing
the `RaggedArray` instance to a `GRangesList`; this is simplified
to a `GRanges` with `unlist`.
```{r geta,eval=FALSE}
gr = unlist(as(mut, "GRangesList"))
dim(mcols(gr))
head(gr[,1:3],3)
Multibase variants are present. We'll confine attention to single-nucleotide variants (SNV). ```{r getsnv, eval=FALSE} table(width(gr))[1:6] gr = gr[width(gr)==1]
We'll want the variants in GRCh38 coordinates.
```{r doma,eval=FALSE}
seqlevels(gr) = paste0("chr", seqlevels(gr))
gr38 = unlist(rtracklayer::liftOver(gr, oc2bioc::ch19to38))
genome(gr38) = "hg38" # UCSC terminology
head(gr38[,1:3],3)
length(gr38)
length(unique(gr38))
Transforming variants to a TSV file for use with R and OpenCRAVAT
```{r mm} library(oc2bioc) mtb = make_oc_POSTable(gr38) # already remapped head(mtb)
Use code like
write.table(mtb, file="/home/rstudio/accdemo.tsv", sep="\t", col.names=FALSE, row.names=FALSE, quote=FALSE)
to place the formatted mutation data where it is easily found.
Now, invoke the shiny app for OpenCRAVAT annotation and reporting:
oc2bioc(".local/bin/cravat") ``` Click on the radio button corresponding to the tsv file created above. The following will eventually appear:
Details on the unique variants reported are found at the variants tab:
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.