In LieberInstitute/spatialLIBD: spatialLIBD: an R/Bioconductor package to visualize spatially-resolved transcriptomics data
knitr::opts_chunk$set(
collapse = TRUE,
comment = "#>",
crop = NULL ## Related to https://stat.ethz.ch/pipermail/bioc-devel/2020-April/016656.html
)
## Track time spent on making the vignette
startTime <- Sys.time()
## Bib setup
library("RefManageR")
## Write bibliography information
bib <- c(
R = citation(),
BiocStyle = citation("BiocStyle")[1],
knitr = citation("knitr")[1],
RefManageR = citation("RefManageR")[1],
rmarkdown = citation("rmarkdown")[1],
sessioninfo = citation("sessioninfo")[1],
testthat = citation("testthat")[1],
spatialLIBD = citation("spatialLIBD")[1],
spatialLIBDpaper = citation("spatialLIBD")[2],
tran2021 = RefManageR::BibEntry(
bibtype = "Article",
key = "tran2021",
author = "Tran, Matthew N. and Maynard, Kristen R. and Spangler, Abby and Huuki, Louise A. and Montgomery, Kelsey D. and Sadashivaiah, Vijay and Tippani, Madhavi and Barry, Brianna K. and Hancock, Dana B. and Hicks, Stephanie C. and Kleinman, Joel E. and Hyde, Thomas M. and Collado-Torres, Leonardo and Jaffe, Andrew E. and Martinowich, Keri",
title = "Single-nucleus transcriptome analysis reveals cell-type-specific molecular signatures across reward circuitry in the human brain",
year = 2021, doi = "10.1016/j.neuron.2021.09.001",
journal = "Neuron"
)
)
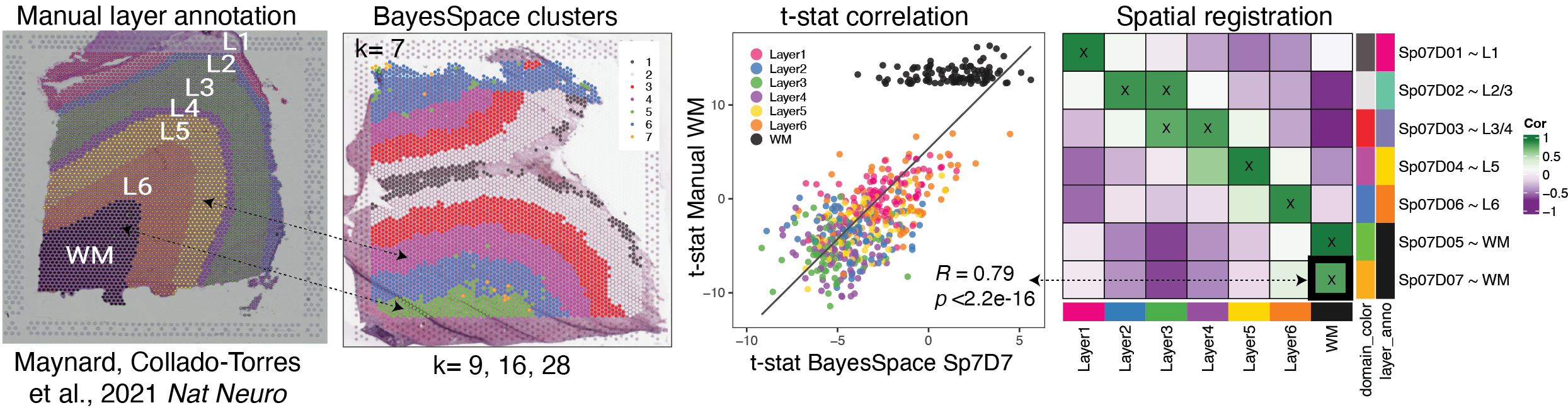
What is Spatial Registration?
Spatial Registration is an analysis that compares the gene expression of groups
in a query RNA-seq data set (typically spatially resolved RNA-seq or single cell RNA-seq) to
groups in a reference spatially resolved RNA-seq data set (such annotated anatomical features).
For spatial data, this can be helpful to compare manual annotations,
or annotating clusters. For scRNA-seq data it can check if
a cell type might be more concentrated in one area or anatomical feature of the
tissue.
The spatial annotation process correlates the $t$-statistics from the gene enrichment
analysis between spatial features from the reference data set, with the $t$-statistics
from the gene enrichment of features in the query data set. Pairs with high
positive correlation show where similar patterns of gene expression are occurring
and what anatomical feature the new spatial feature or cell population may map to.
Overview of the Spatial Registration method
-
Perform gene set enrichment analysis between spatial features (ex. anatomical
features, histological layers) on reference spatial data set. Or access existing statistics.
-
Perform gene set enrichment analysis between features (ex. new
annotations, data-driven clusters) on new query data set.
-
Correlate the $t$-statistics between the reference and query features.
-
Annotate new spatial features with the most strongly associated reference feature.
-
Plot correlation heat map to observe patterns between the two data sets.
{width=100%}
How to run Spatial Registration with spatialLIBD tools
Introduction.
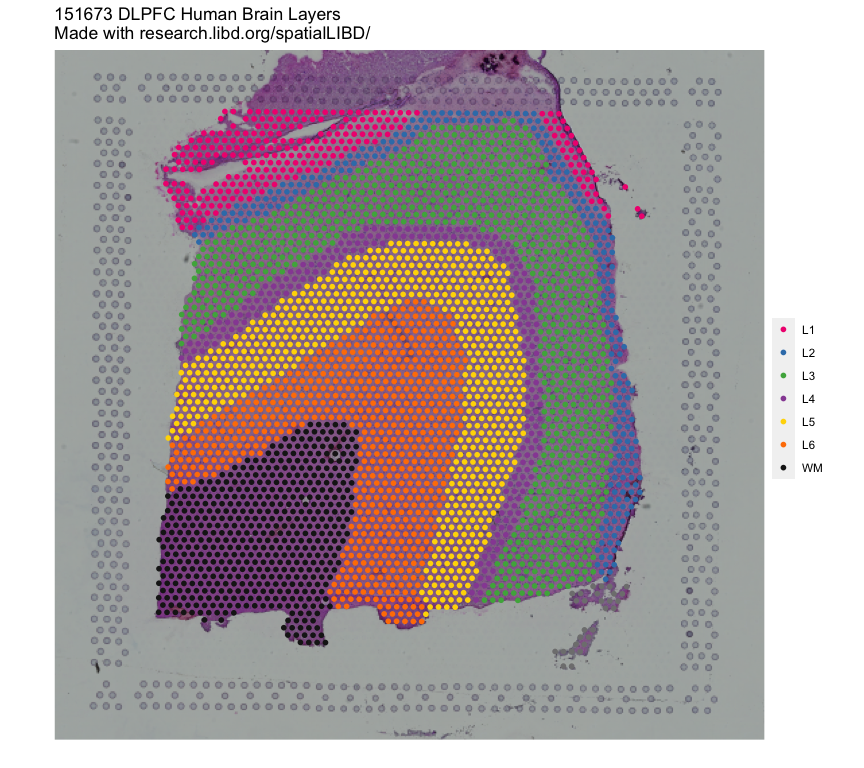
In this example we will utilize the human DLPFC 10x Genomics Visium dataset
from Maynard, Collado-Torres et al. r Citep(bib[['spatialLIBDpaper']]) as the reference.
This data contains manually annotated features: the six cortical layers + white matter
present in the DLPFC. We will use the pre-calculated enrichment statistics for the
layers, which are available from r Biocpkg("spatialLIBD").
{width=100%}
The query dataset will be the DLPFC single nucleus RNA-seq (snRNA-seq) data from r Citep(bib[['tran2021']]).
We will compare the gene expression in the cell type populations of the query
dataset to the annotated layers in the reference.
Important Notes
Required knowledge
It may be helpful to review Introduction to spatialLIBD vignette available through GitHub or Bioconductor for more information about this data set and R package.
Citing spatialLIBD
We hope that r Biocpkg("spatialLIBD") will be useful for your research. Please use the following information to cite the package and the overall approach. Thank you!
## Citation info
citation("spatialLIBD")
Setup
Install spatialLIBD
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("spatialLIBD")
## Check that you have a valid Bioconductor installation
BiocManager::valid()
Load required packages
library("spatialLIBD")
library("SingleCellExperiment")
Download Data
Spatial Reference
The reference data is easily accessed through r Biocpkg("spatialLIBD"). The modeling results
for the annotated layers is already calculated and can be accessed with the fetch_data() function.
This data contains the results form three models (anova, enrichment, and pairwise),
we will use the enrichment results for spatial registration. The tables contain the
$t$-statistics, p-values, and gene ensembl ID and symbol.
## get reference layer enrichment statistics
layer_modeling_results <- fetch_data(type = "modeling_results")
layer_modeling_results$enrichment[1:5, 1:5]
Query Data: snRNA-seq
For the query data set, we will use the public single nucleus RNA-seq (snRNA-seq)
data from r Citep(bib[['tran2021']]) can be accessed on github.
This data is also from postmortem human brain DLPFC, and contains gene
expression data for 11k nuclei and 19 cell types.
We will use BiocFileCache() to cache this data. It is stored as a SingleCellExperiment
object named sce.dlpfc.tran, and takes 1.01 GB of RAM memory to load.
# Download and save a local cache of the data available at:
# https://github.com/LieberInstitute/10xPilot_snRNAseq-human#processed-data
bfc <- BiocFileCache::BiocFileCache()
url <- paste0(
"https://libd-snrnaseq-pilot.s3.us-east-2.amazonaws.com/",
"SCE_DLPFC-n3_tran-etal.rda"
)
local_data <- BiocFileCache::bfcrpath(url, x = bfc)
load(local_data, verbose = TRUE)
DLPFC tissue consists of many cell types, some are quite rare and will not have enough data to complete the analysis
table(sce.dlpfc.tran$cellType)
The data will be pseudo-bulked over donor x cellType, it is recommended to drop
groups with < 10 nuclei (this is done automatically in the pseudobulk step).
table(sce.dlpfc.tran$donor, sce.dlpfc.tran$cellType)
Get Enrichment statistics for snRNA-seq data
spatialLIBD contains many functions to compute modeling_results for the query sc/snRNA-seq or spatial data.
The process includes the following steps
registration_pseudobulk(): Pseudo-bulks data, filter low expressed genes, and normalize countsregistration_mod(): Defines the statistical model that will be used for computing the block correlationregistration_block_cor() : Computes the block correlation using the sample ID as the blocking factor, used as correlation in eBayes callregistration_stats_enrichment() : Computes the gene enrichment $t$-statistics (one group vs. All other groups)
The function registration_wrapper() makes life easy by wrapping these functions together in to one step!
## Perform the spatial registration
sce_modeling_results <- registration_wrapper(
sce = sce.dlpfc.tran,
var_registration = "cellType",
var_sample_id = "donor",
gene_ensembl = "gene_id",
gene_name = "gene_name"
)
Extract Enrichment t-statistics
## check out table on enrichment t-statistics
sce_modeling_results$enrichment[1:5, 1:5]
Correlate statsics with Layer Reference
cor_layer <- layer_stat_cor(
stats = sce_modeling_results$enrichment,
modeling_results = layer_modeling_results,
model_type = "enrichment",
top_n = 100
)
cor_layer
Explore Results
Now we can use these correlation values to learn about the cell types.
Create Heatmap of Correlations
We can see from this heatmap what layers the different cell types are associated with.
-
Oligo with WM
-
Astro with Layer 1
-
Excitatory neurons to different layers of the cortex
-
Weak associate with Inhibitory Neurons
layer_stat_cor_plot(cor_layer)
Annotate Cell Types by Top Correlation
We can use annotate_registered_clusters to create annotation labels for the
cell types based on the correlation values.
anno <- annotate_registered_clusters(
cor_stats_layer = cor_layer,
confidence_threshold = 0.25,
cutoff_merge_ratio = 0.25
)
anno
Plot Annotated Cell Types
Finally, we can update our heatmap with colors and annotations based on cluster
registration for the snRNA-seq clusters.
layer_stat_cor_plot(
cor_layer,
query_colors = get_colors(clusters = rownames(cor_layer)),
reference_colors = libd_layer_colors,
annotation = anno,
cluster_rows = FALSE,
cluster_columns = FALSE
)
Reproducibility
The r Biocpkg("spatialLIBD") package r Citep(bib[["spatialLIBD"]]) was made possible thanks to:
- R
r Citep(bib[["R"]])
r Biocpkg("BiocStyle") r Citep(bib[["BiocStyle"]])r CRANpkg("knitr") r Citep(bib[["knitr"]])r CRANpkg("RefManageR") r Citep(bib[["RefManageR"]])r CRANpkg("rmarkdown") r Citep(bib[["rmarkdown"]])r CRANpkg("sessioninfo") r Citep(bib[["sessioninfo"]])r CRANpkg("testthat") r Citep(bib[["testthat"]])
This package was developed using r BiocStyle::Biocpkg("biocthis").
Code for creating the vignette
## Create the vignette
library("rmarkdown")
system.time(render("guide_to_spatial_registration.Rmd", "BiocStyle::html_document"))
## Extract the R code
library("knitr")
knit("guide_to_spatial_registration.Rmd", tangle = TRUE)
Date the vignette was generated.
## Date the vignette was generated
Sys.time()
Wallclock time spent generating the vignette.
## Processing time in seconds
totalTime <- diff(c(startTime, Sys.time()))
round(totalTime, digits = 3)
R session information.
## Session info
library("sessioninfo")
options(width = 120)
session_info()
Bibliography
This vignette was generated using r Biocpkg("BiocStyle") r Citep(bib[["BiocStyle"]])
with r CRANpkg("knitr") r Citep(bib[["knitr"]]) and r CRANpkg("rmarkdown") r Citep(bib[["rmarkdown"]]) running behind the scenes.
Citations made with r CRANpkg("RefManageR") r Citep(bib[["RefManageR"]]).
## Print bibliography
PrintBibliography(bib, .opts = list(hyperlink = "to.doc", style = "html"))
LieberInstitute/spatialLIBD documentation built on April 14, 2025, 5:19 a.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
knitr::opts_chunk$set( collapse = TRUE, comment = "#>", crop = NULL ## Related to https://stat.ethz.ch/pipermail/bioc-devel/2020-April/016656.html )
## Track time spent on making the vignette startTime <- Sys.time() ## Bib setup library("RefManageR") ## Write bibliography information bib <- c( R = citation(), BiocStyle = citation("BiocStyle")[1], knitr = citation("knitr")[1], RefManageR = citation("RefManageR")[1], rmarkdown = citation("rmarkdown")[1], sessioninfo = citation("sessioninfo")[1], testthat = citation("testthat")[1], spatialLIBD = citation("spatialLIBD")[1], spatialLIBDpaper = citation("spatialLIBD")[2], tran2021 = RefManageR::BibEntry( bibtype = "Article", key = "tran2021", author = "Tran, Matthew N. and Maynard, Kristen R. and Spangler, Abby and Huuki, Louise A. and Montgomery, Kelsey D. and Sadashivaiah, Vijay and Tippani, Madhavi and Barry, Brianna K. and Hancock, Dana B. and Hicks, Stephanie C. and Kleinman, Joel E. and Hyde, Thomas M. and Collado-Torres, Leonardo and Jaffe, Andrew E. and Martinowich, Keri", title = "Single-nucleus transcriptome analysis reveals cell-type-specific molecular signatures across reward circuitry in the human brain", year = 2021, doi = "10.1016/j.neuron.2021.09.001", journal = "Neuron" ) )
What is Spatial Registration?
Spatial Registration is an analysis that compares the gene expression of groups in a query RNA-seq data set (typically spatially resolved RNA-seq or single cell RNA-seq) to groups in a reference spatially resolved RNA-seq data set (such annotated anatomical features).
For spatial data, this can be helpful to compare manual annotations, or annotating clusters. For scRNA-seq data it can check if a cell type might be more concentrated in one area or anatomical feature of the tissue.
The spatial annotation process correlates the $t$-statistics from the gene enrichment analysis between spatial features from the reference data set, with the $t$-statistics from the gene enrichment of features in the query data set. Pairs with high positive correlation show where similar patterns of gene expression are occurring and what anatomical feature the new spatial feature or cell population may map to.
Overview of the Spatial Registration method
-
Perform gene set enrichment analysis between spatial features (ex. anatomical features, histological layers) on reference spatial data set. Or access existing statistics.
-
Perform gene set enrichment analysis between features (ex. new annotations, data-driven clusters) on new query data set.
-
Correlate the $t$-statistics between the reference and query features.
-
Annotate new spatial features with the most strongly associated reference feature.
-
Plot correlation heat map to observe patterns between the two data sets.
{width=100%}
How to run Spatial Registration with spatialLIBD tools
Introduction.
In this example we will utilize the human DLPFC 10x Genomics Visium dataset
from Maynard, Collado-Torres et al. r Citep(bib[['spatialLIBDpaper']]) as the reference.
This data contains manually annotated features: the six cortical layers + white matter
present in the DLPFC. We will use the pre-calculated enrichment statistics for the
layers, which are available from r Biocpkg("spatialLIBD").
{width=100%}
The query dataset will be the DLPFC single nucleus RNA-seq (snRNA-seq) data from r Citep(bib[['tran2021']]).
We will compare the gene expression in the cell type populations of the query dataset to the annotated layers in the reference.
Important Notes
Required knowledge
It may be helpful to review Introduction to spatialLIBD vignette available through GitHub or Bioconductor for more information about this data set and R package.
Citing spatialLIBD
We hope that r Biocpkg("spatialLIBD") will be useful for your research. Please use the following information to cite the package and the overall approach. Thank you!
## Citation info citation("spatialLIBD")
Setup
Install spatialLIBD
if (!requireNamespace("BiocManager", quietly = TRUE)) { install.packages("BiocManager") } BiocManager::install("spatialLIBD") ## Check that you have a valid Bioconductor installation BiocManager::valid()
Load required packages
library("spatialLIBD") library("SingleCellExperiment")
Download Data
Spatial Reference
The reference data is easily accessed through r Biocpkg("spatialLIBD"). The modeling results
for the annotated layers is already calculated and can be accessed with the fetch_data() function.
This data contains the results form three models (anova, enrichment, and pairwise), we will use the enrichment results for spatial registration. The tables contain the $t$-statistics, p-values, and gene ensembl ID and symbol.
## get reference layer enrichment statistics layer_modeling_results <- fetch_data(type = "modeling_results") layer_modeling_results$enrichment[1:5, 1:5]
Query Data: snRNA-seq
For the query data set, we will use the public single nucleus RNA-seq (snRNA-seq)
data from r Citep(bib[['tran2021']]) can be accessed on github.
This data is also from postmortem human brain DLPFC, and contains gene expression data for 11k nuclei and 19 cell types.
We will use BiocFileCache() to cache this data. It is stored as a SingleCellExperiment
object named sce.dlpfc.tran, and takes 1.01 GB of RAM memory to load.
# Download and save a local cache of the data available at: # https://github.com/LieberInstitute/10xPilot_snRNAseq-human#processed-data bfc <- BiocFileCache::BiocFileCache() url <- paste0( "https://libd-snrnaseq-pilot.s3.us-east-2.amazonaws.com/", "SCE_DLPFC-n3_tran-etal.rda" ) local_data <- BiocFileCache::bfcrpath(url, x = bfc) load(local_data, verbose = TRUE)
DLPFC tissue consists of many cell types, some are quite rare and will not have enough data to complete the analysis
table(sce.dlpfc.tran$cellType)
The data will be pseudo-bulked over donor x cellType, it is recommended to drop
groups with < 10 nuclei (this is done automatically in the pseudobulk step).
table(sce.dlpfc.tran$donor, sce.dlpfc.tran$cellType)
Get Enrichment statistics for snRNA-seq data
spatialLIBD contains many functions to compute modeling_results for the query sc/snRNA-seq or spatial data.
The process includes the following steps
registration_pseudobulk(): Pseudo-bulks data, filter low expressed genes, and normalize countsregistration_mod(): Defines the statistical model that will be used for computing the block correlationregistration_block_cor(): Computes the block correlation using the sample ID as the blocking factor, used as correlation in eBayes callregistration_stats_enrichment(): Computes the gene enrichment $t$-statistics (one group vs. All other groups)
The function registration_wrapper() makes life easy by wrapping these functions together in to one step!
## Perform the spatial registration sce_modeling_results <- registration_wrapper( sce = sce.dlpfc.tran, var_registration = "cellType", var_sample_id = "donor", gene_ensembl = "gene_id", gene_name = "gene_name" )
Extract Enrichment t-statistics
## check out table on enrichment t-statistics sce_modeling_results$enrichment[1:5, 1:5]
Correlate statsics with Layer Reference
cor_layer <- layer_stat_cor( stats = sce_modeling_results$enrichment, modeling_results = layer_modeling_results, model_type = "enrichment", top_n = 100 ) cor_layer
Explore Results
Now we can use these correlation values to learn about the cell types.
Create Heatmap of Correlations
We can see from this heatmap what layers the different cell types are associated with.
-
Oligo with WM
-
Astro with Layer 1
-
Excitatory neurons to different layers of the cortex
-
Weak associate with Inhibitory Neurons
layer_stat_cor_plot(cor_layer)
Annotate Cell Types by Top Correlation
We can use annotate_registered_clusters to create annotation labels for the
cell types based on the correlation values.
anno <- annotate_registered_clusters( cor_stats_layer = cor_layer, confidence_threshold = 0.25, cutoff_merge_ratio = 0.25 ) anno
Plot Annotated Cell Types
Finally, we can update our heatmap with colors and annotations based on cluster registration for the snRNA-seq clusters.
layer_stat_cor_plot( cor_layer, query_colors = get_colors(clusters = rownames(cor_layer)), reference_colors = libd_layer_colors, annotation = anno, cluster_rows = FALSE, cluster_columns = FALSE )
Reproducibility
The r Biocpkg("spatialLIBD") package r Citep(bib[["spatialLIBD"]]) was made possible thanks to:
- R
r Citep(bib[["R"]]) r Biocpkg("BiocStyle")r Citep(bib[["BiocStyle"]])r CRANpkg("knitr")r Citep(bib[["knitr"]])r CRANpkg("RefManageR")r Citep(bib[["RefManageR"]])r CRANpkg("rmarkdown")r Citep(bib[["rmarkdown"]])r CRANpkg("sessioninfo")r Citep(bib[["sessioninfo"]])r CRANpkg("testthat")r Citep(bib[["testthat"]])
This package was developed using r BiocStyle::Biocpkg("biocthis").
Code for creating the vignette
## Create the vignette library("rmarkdown") system.time(render("guide_to_spatial_registration.Rmd", "BiocStyle::html_document")) ## Extract the R code library("knitr") knit("guide_to_spatial_registration.Rmd", tangle = TRUE)
Date the vignette was generated.
## Date the vignette was generated Sys.time()
Wallclock time spent generating the vignette.
## Processing time in seconds totalTime <- diff(c(startTime, Sys.time())) round(totalTime, digits = 3)
R session information.
## Session info library("sessioninfo") options(width = 120) session_info()
Bibliography
This vignette was generated using r Biocpkg("BiocStyle") r Citep(bib[["BiocStyle"]])
with r CRANpkg("knitr") r Citep(bib[["knitr"]]) and r CRANpkg("rmarkdown") r Citep(bib[["rmarkdown"]]) running behind the scenes.
Citations made with r CRANpkg("RefManageR") r Citep(bib[["RefManageR"]]).
## Print bibliography PrintBibliography(bib, .opts = list(hyperlink = "to.doc", style = "html"))
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.