README.md
In Pitithat-pu/cfdnakit: Fragmen-length analysis package from high-throughput sequencing of cell-free DNA (cfDNA)
cfdnakit : an R package for fragmentation analysis of cfDNA and estimation of circulating tumor DNA from NGS data.
Installation
Install via the Bioconductor repository
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("cfdnakit")
Install the latest version via github
Package was tested on R environment 4.0.0. To install this package is via this github repository. please follow instruction below.
Install prerequisites packages
if(! "devtools" %in% rownames(installed.packages()))
install.packages("devtools")
if(! "BiocManager" %in% rownames(installed.packages()))
install.packages("BiocManager")
Install cfdnakit package
library(devtools) ### use devtools
install_github("Pitithat-pu/cfdnakit") ### install cfDNAKit
The installation should work fine without non-zero exit status. Try load cfdnakit package into current R session
library(cfdnakit) ### Load cfdnakit package
Usage
Please follow the instructions on GitHub Wiki page
Introduction
This package provides basic functions for analyzing next-generation sequencing of circulating cell-free DNA (cfDNA). The package focuses on extracting length of cfDNA, and genome-wide copy-number alteration estimated by the short-fragmented cfDNA using shallow whole-genome sequencing data (\~0.3X or more). The ctDNA estimation score (CES) comprehensively estimate the circulating tumor DNA based on the short-fragment analysis.
Overview
The figure below shows the overview of the analysis procedure possible within this package. The amount of short-fragmented cfDNA per non-overlapping genomics windows are normalized and compared to a Panel-of-Normal (control). Segmentation is performed using the PSCBS package. A CES score (adapted from Raman, Lennart, et al. 2020) were calculated to estimate circulating tumor dna in the given sample.
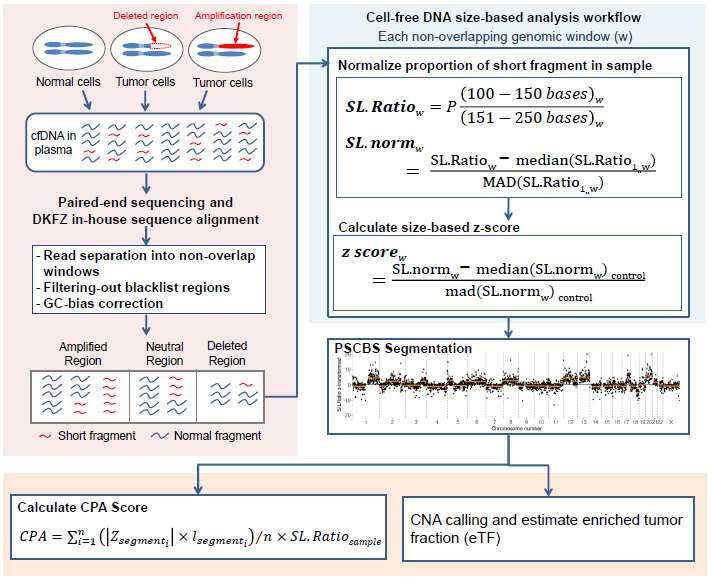
Available functions
The scope of this R package is to analyse the length of cfDNA fragments. The package simplifies the process of extracting length of fragments from a BAM file and provides basic functions to explore this characteristic of cfDNA with low-coverage whole-genome sequencing data. Moreover, this package utilizes the quantity of short-fragmented cfDNA to infer copy-number alterations and estimate the percentage of tumor-derived cfDNA.
Excluding ENCODE blacklist loci
It is recommended when analyzing genomic data to exclude sequencing reads locate within the ENCODE blacklist loci to assure the quality of the result. When using the GRCh37 as the reference in cfdnakit, a set of genomic regions including the ENCODE blacklist and centromere loci, provided by UCSC Genome Browser, were used. Users can introduce customized blacklist regions by creating a bed file or a tab-separated file where the first three columns are chromosome, start, and end position respectively. The future cfdnakit would be able to support blacklists of other reference genomes such as GRCh38 or GRCm38.
GC and Mappability Bias Correction
LOESS regression model is created from the relation between the fragment count and the percent of GC per bin. The raw count per bin is deduced with the read count predicted by the model. Then, the values are added with the median of raw counts to bring back the range of values similar to the raw count. After correction for GC bias, the GC-corrected read counts are then corrected for mappability bias, capability of a genomic region to be mapped uniquely by sequencing reads, using a similar process.
Fragment length distribution and comparison
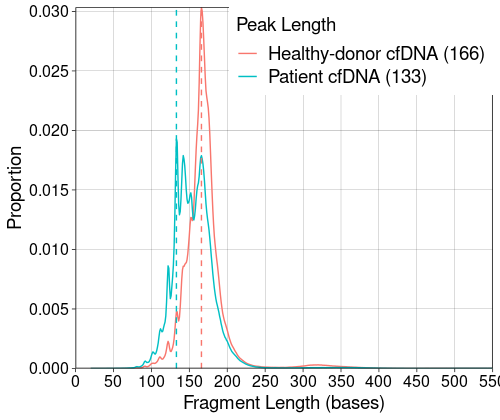
Package provides a single function to extract fragment length of cfDNA in the sample. Making a fragment-length distribution plot of multiple samples is easy. cfdnakit also extracted the short-fragment ratio representing the amount of short-fragmented cfdNA in the sample. It can be used for comparison between groups of sample (e.g. healthy vs patient) or for quality control inspection.
This plot shows the fragment-length distribution of cfDNA from a healthy individual cfDNA (red) and a patient-derived cfDNA (blue). The top-right legend report the leak length per individual sample as shown.
Sample Short-fragmented Ratio
An enrichment of short-fragmented cfDNA (\<150 base) were commonly found in tumor-derived cfDNA. This package extracts a preliminary estimation of short-fragmented cfDNA by calculating Short-fragmented Ratio (S.L.Ratio) inferring to the proportion of short-fragmented cfDNA (100 to 150 by default) over long-fragmented cfDNA (151 to 250 by default).

where  is number of short fragments;
is number of short fragments;  is number of long fragments;
is number of long fragments;
 can be used as a general quantification of ctDNA for comparing plasma cfDNA samples. This ratio increases when a sample contains the higher contribution of ctDNA.
can be used as a general quantification of ctDNA for comparing plasma cfDNA samples. This ratio increases when a sample contains the higher contribution of ctDNA.
Genome-wide Short-fragmented Ratio
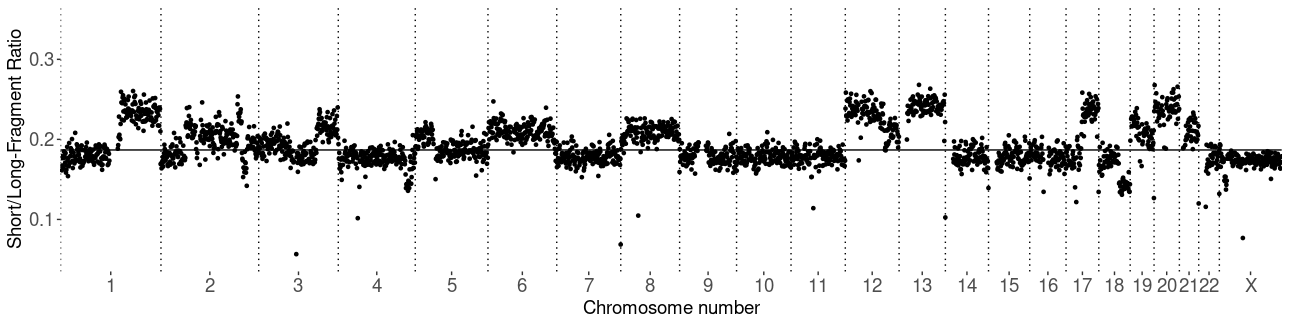
The proportion of short-fragmented cfDNA is positively correlated with copy-number aberration. It shows that a short-fragment ratio of a genomic segment is increasing in the amplified segments and decreasing when the segment is lost.
Cfdnakit package separates input reads into non-overlapping bins with equal size (1 MB by default) and calculated S.L.Ratio. The S.L.Ratio per bin is plotted in genomic order as followed.
Normalized with a Panel-of-normal and segmentation
To estimate the rate of both technical and biological artifacts, creation of a Panel-of-Normal (PoN) is usually recommended by most bioinformatics workflow. A PoN of cfDNA analysis should be made from healthy samples or a group of selected patient-derived cfDNA. There is no definitive rule on how to select or how many samples should be included in a PoN. Creating a PoN will in general be better than analysis without a PoN. Nevertheless, the most important approach is including normal samples that are generated by similar techniques (such as DNA preparation methods, sequencing platform, and biological sources) as many as possible.
Cfdnakit transforms S.L.Ratio per bin into zscore by subtracting the median and dividing by median absolute deviation (MAD). Finally, cfdnakit perform Circular Binary Segmentation (CBS) implemented in PSCBS. The result of transformation and segmentation can be plotted as followed.

Modified copy number profile abnormality score
Cfdnakit calculates ctDNA estimation score (CES) from the result of segmentation and sample S.L.Ratio. which robust to coverage bias and noisy fragmented signals.The score quantify the overall genomic aberration of short-fragmented cfDNA. The higher score infering to enrichment of short-fragmented cfDNA in the sample and copy-number aberrations.
Contact
If you have any questions or feedback, please contact us at: Email: pitithat.pur\@cra.ac.th; b.brors\@dkfz-heidelberg.de
Pitithat-pu/cfdnakit documentation built on Oct. 2, 2024, 8:03 p.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
cfdnakit : an R package for fragmentation analysis of cfDNA and estimation of circulating tumor DNA from NGS data.
Installation
Install via the Bioconductor repository
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("cfdnakit")
Install the latest version via github
Package was tested on R environment 4.0.0. To install this package is via this github repository. please follow instruction below.
Install prerequisites packages
if(! "devtools" %in% rownames(installed.packages()))
install.packages("devtools")
if(! "BiocManager" %in% rownames(installed.packages()))
install.packages("BiocManager")
Install cfdnakit package
library(devtools) ### use devtools
install_github("Pitithat-pu/cfdnakit") ### install cfDNAKit
The installation should work fine without non-zero exit status. Try load cfdnakit package into current R session
library(cfdnakit) ### Load cfdnakit package
Usage
Please follow the instructions on GitHub Wiki page
Introduction
This package provides basic functions for analyzing next-generation sequencing of circulating cell-free DNA (cfDNA). The package focuses on extracting length of cfDNA, and genome-wide copy-number alteration estimated by the short-fragmented cfDNA using shallow whole-genome sequencing data (\~0.3X or more). The ctDNA estimation score (CES) comprehensively estimate the circulating tumor DNA based on the short-fragment analysis.
Overview
The figure below shows the overview of the analysis procedure possible within this package. The amount of short-fragmented cfDNA per non-overlapping genomics windows are normalized and compared to a Panel-of-Normal (control). Segmentation is performed using the PSCBS package. A CES score (adapted from Raman, Lennart, et al. 2020) were calculated to estimate circulating tumor dna in the given sample.
Available functions
The scope of this R package is to analyse the length of cfDNA fragments. The package simplifies the process of extracting length of fragments from a BAM file and provides basic functions to explore this characteristic of cfDNA with low-coverage whole-genome sequencing data. Moreover, this package utilizes the quantity of short-fragmented cfDNA to infer copy-number alterations and estimate the percentage of tumor-derived cfDNA.
Excluding ENCODE blacklist loci
It is recommended when analyzing genomic data to exclude sequencing reads locate within the ENCODE blacklist loci to assure the quality of the result. When using the GRCh37 as the reference in cfdnakit, a set of genomic regions including the ENCODE blacklist and centromere loci, provided by UCSC Genome Browser, were used. Users can introduce customized blacklist regions by creating a bed file or a tab-separated file where the first three columns are chromosome, start, and end position respectively. The future cfdnakit would be able to support blacklists of other reference genomes such as GRCh38 or GRCm38.
GC and Mappability Bias Correction
LOESS regression model is created from the relation between the fragment count and the percent of GC per bin. The raw count per bin is deduced with the read count predicted by the model. Then, the values are added with the median of raw counts to bring back the range of values similar to the raw count. After correction for GC bias, the GC-corrected read counts are then corrected for mappability bias, capability of a genomic region to be mapped uniquely by sequencing reads, using a similar process.
Fragment length distribution and comparison
Package provides a single function to extract fragment length of cfDNA in the sample. Making a fragment-length distribution plot of multiple samples is easy. cfdnakit also extracted the short-fragment ratio representing the amount of short-fragmented cfdNA in the sample. It can be used for comparison between groups of sample (e.g. healthy vs patient) or for quality control inspection.
This plot shows the fragment-length distribution of cfDNA from a healthy individual cfDNA (red) and a patient-derived cfDNA (blue). The top-right legend report the leak length per individual sample as shown.
Sample Short-fragmented Ratio
An enrichment of short-fragmented cfDNA (\<150 base) were commonly found in tumor-derived cfDNA. This package extracts a preliminary estimation of short-fragmented cfDNA by calculating Short-fragmented Ratio (S.L.Ratio) inferring to the proportion of short-fragmented cfDNA (100 to 150 by default) over long-fragmented cfDNA (151 to 250 by default).
where is number of short fragments;
is number of long fragments;
can be used as a general quantification of ctDNA for comparing plasma cfDNA samples. This ratio increases when a sample contains the higher contribution of ctDNA.
Genome-wide Short-fragmented Ratio
The proportion of short-fragmented cfDNA is positively correlated with copy-number aberration. It shows that a short-fragment ratio of a genomic segment is increasing in the amplified segments and decreasing when the segment is lost.
Cfdnakit package separates input reads into non-overlapping bins with equal size (1 MB by default) and calculated S.L.Ratio. The S.L.Ratio per bin is plotted in genomic order as followed.
Normalized with a Panel-of-normal and segmentation
To estimate the rate of both technical and biological artifacts, creation of a Panel-of-Normal (PoN) is usually recommended by most bioinformatics workflow. A PoN of cfDNA analysis should be made from healthy samples or a group of selected patient-derived cfDNA. There is no definitive rule on how to select or how many samples should be included in a PoN. Creating a PoN will in general be better than analysis without a PoN. Nevertheless, the most important approach is including normal samples that are generated by similar techniques (such as DNA preparation methods, sequencing platform, and biological sources) as many as possible.
Cfdnakit transforms S.L.Ratio per bin into zscore by subtracting the median and dividing by median absolute deviation (MAD). Finally, cfdnakit perform Circular Binary Segmentation (CBS) implemented in PSCBS. The result of transformation and segmentation can be plotted as followed.
![]()
Modified copy number profile abnormality score
Cfdnakit calculates ctDNA estimation score (CES) from the result of segmentation and sample S.L.Ratio. which robust to coverage bias and noisy fragmented signals.The score quantify the overall genomic aberration of short-fragmented cfDNA. The higher score infering to enrichment of short-fragmented cfDNA in the sample and copy-number aberrations.
Contact
If you have any questions or feedback, please contact us at: Email: pitithat.pur\@cra.ac.th; b.brors\@dkfz-heidelberg.de
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.