In famuvie/breedR: Statistical Methods for Forest Genetic Resources Analysts
library(knitr)
opts_chunk$set(echo = TRUE,
message = TRUE,
warning = TRUE,
# comment = NA,
fig.width = 4,
fig.height = 3,
cache = TRUE,
autodep = TRUE,
## invalidate cache when either versions of R or breedR change
## http://yihui.name/knitr/demo/cache/
cache.extra= list(R.version, packageVersion('breedR')))
library(breedR)
library(ggplot2)
Intro
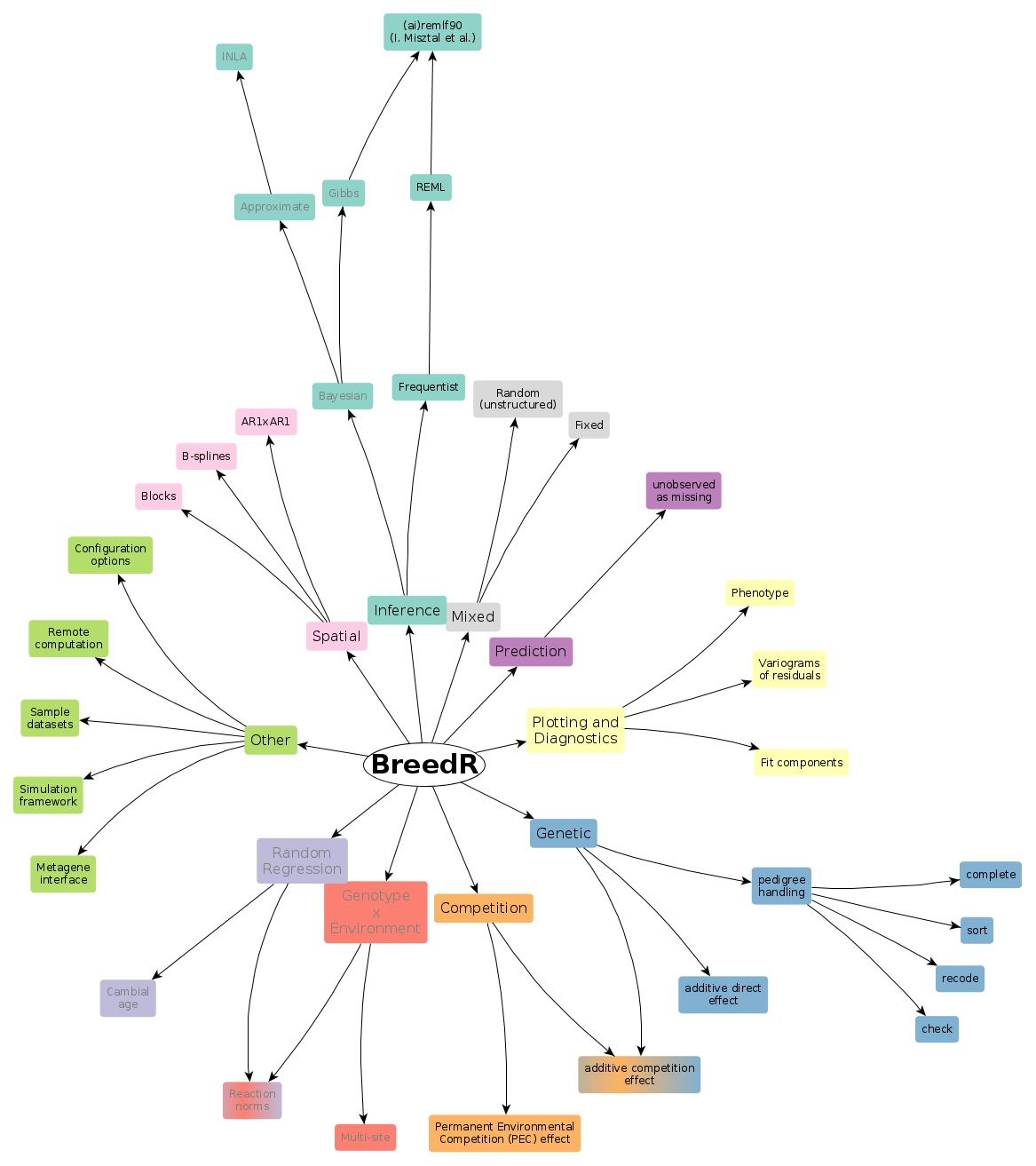
What is breedR
- R-package implementing statistical models specifically suited for forest genetic resources analysts.
- Ultimately Mixed Models, but not necessarily easy to implement and use
-
breedR acts as an interface which provides the means to:
- Combine any number of these models as components of a larger model
- Compute automatically incidence and covariance matrices from a few input parameters
- Fit the model
- Plot data and results, and perform model diagnostics
Installation
-
Project web page http://famuvie.github.io/breedR/
- Set up this URL as a package repository in
.Rprofile (detailed instructions on the web)
install.packages('breedR')- Not possible to use
CRAN due to closed-source BLUPF90 programs
-
GitHub dev-site https://github.com/famuvie/breedR
if( !require(devtools) ) install.packages('devtools')devtools::install_github('famuvie/breedR')
Where to find help
-
Package's help: help(package = breedR)
- Help pages
?remlf90
- Code demos
demo(topic, package = 'breedR') (omit topic for a list)
- Vignettes
vignette(package = 'breedR') (pkg and wiki)
-
- Guides, tutorials, FAQ
-
Mailing list http://groups.google.com/group/breedr
- Questions and debates about usage and interface
-
- Bug reports
- Feature requests
License

- breedR is FOSS. Licensed GPL-3
RShowDoc('LICENSE', package = 'breedR')
- You can use and distribute breedR for any purpose
-
You can modify it to suit your needs
- we encourage to!
- please consider contributing your improvements
- you can distribute your modified version under the GPL
-
However, breedR makes (intensive) use of the BLUPF90 suite of Fortran programs, which are for free but not free (remember CRAN?)
Roadmap | Future developments
-
Bayesian inference
-
Multi-trait support
-
Genotype$\times$Environment interaction
-
Support for longitudinal data
Functionality

Inference
Frequentist
-
Currently, only frequentist inference is supported via REML estimation of variance components.
-
The function remlf90(), provides an interface to both REMLF90 and AIREMLF90 functions in the BLUPF90 suite of Fortran programs.
-
Type ?remlf90 for details on the syntax
Bayesian
-
It's on the roadmap for the next year
-
Will use a gibbs sampler from BLUPF90, and possibly also INLA
-
The interface will change a bit, separating the model specification from the fit
Linear Mixed Models with unstructured random effects
Example dataset
knitr::kable(head(globulus))
cat(attr(globulus, 'comment'), sep ='\n')
str(globulus, give.attr = FALSE)
A simple Provenance Test
Specify the genetic group gg as an unstructured random effect using the standard formulas in R
$$
\begin{aligned}
\mathrm{phe}X = & \mu + Z \mathrm{gg} + \varepsilon \
\mathrm{gg} \sim & N(0, \sigma{\mathrm{gg}}^2) \
\varepsilon \sim & N(0, \sigma_\varepsilon^2)
\end{aligned}
$$
res <- remlf90(fixed = phe_X ~ 1,
random = ~ gg,
data = globulus)
Initial variances specification
To avoid the notification, initial values for all the variance components must be made explicit using the argument var.ini:
res <- remlf90(fixed = phe_X ~ 1,
random = ~ gg,
var.ini = list(gg = 2, resid = 10),
data = globulus)
Although in most cases the results will not change at all, we encourage to give explicit initial values for variance components.
Specially when some estimate can be artifact.
This is also useful for checking sensitivity to initial values.
Exploring the results
summary(res)
- Note that
AI-REML has been used by default.
- You can also specify
method = 'em'.
- Learn about the difference.
Further extractor functions
fixef(res)
ranef(res)
Further extractor functions
qplot(
fitted(res),
globulus$phe_X) +
geom_abline(intercept = 0,
slope = 1,
col = 'darkgrey')
str(resid(res))
extractAIC(res)
logLik(res)
Hierarchical and Factorial models
-
In globulus, the family (mum) is nested within the provenance (gg)
-
This is a matter of codification:
Nested factors
|gg | mum |
|---:|:----|
|A | 1 |
|A | 2 |
|B | 3 |
|B | 4 |
Crossed factors
|gg | mum |
|---:|:----|
|A | 1 |
|A | 2 |
|B | 1 |
|B | 2 |
Model specification
- Otherwise, in both cases we specify the model in the same way:
random = ~ gg + factor(mum) # note that mum is numeric
- Furthermore, this approach can handle unbalanced and mixed designs
Interactions
- Standard
R notation:
random = ~ gg * factor(mum)
-
Not available yet (feature request?)
-
Workaround: build the interaction variable manually
-
Example: gg and block are crossed factors
dat <- transform(globulus,
interaction = factor(gg:bl))
random = ~ gg + bl + interaction
Exercise | Hierarchical and Factorial models
-
Use remlf90() and the globulus dataset to fit
- a hierarchical model using
mum within gg
- a factorial model using
gg and bl
-
Explore the results with summary()
- is the family (
mum) effect relevant?
- is there any evidence of interaction between
gg and bl?
Hierarchical and Factorial models #1 | Fitting models
res.h <- remlf90(fixed = phe_X ~ 1,
random = ~ factor(mum) + gg,
data = globulus)
# Interaction variable
globulus.f <- transform(globulus,
gg_bl = factor(gg:bl))
res.f <- remlf90(fixed = phe_X ~ 1,
random = ~ gg + bl + gg_bl,
data = globulus.f)
Hierarchical and Factorial models #2 | Hierarchical model
- The family effect is not very important, in terms of explained variance
- However, the model is a bit better with it (AIC, logLik)
summary(res)
summary(res.h)
Hierarchical and Factorial models #3 | Factorial model
- Looks like the interaction between block and provenance is negligible
- (apart from the fact that it makes no sense at all, and shuld not have been even considered in the first place)
- compare with the model without interaction
summary(res.f)
## result without interaction
res.f0 <- remlf90(fixed = phe_X ~ 1,
random = ~ gg + bl,
data = globulus)
paste('AIC:', round(extractAIC(res.f0)),
'logLik:', round(logLik(res.f0)))
Additive Genetic Effect

What is an additive genetic effect
-
Random effect at individual level
-
Based on a pedigree
-
BLUP of Breeding Values from own and relatives' phenotypes
-
Represents the additive component of the genetic value
-
More general:
- family effect is a particular case
- accounts for more than one generation
- mixed relationships
-
More flexible: allows to select individuals within families
Specifying a pedigree
-
A 3-column data.frame or matrix with the codes for each individual and its parents
-
A family effect is easily translated into a pedigree:
- use the family code as the identification of a fictitious mother
- use
0 or NA as codes for the unknown fathers
knitr::kable(head(globulus[, 1:3]))
Fitting an animal model
res.animal <- remlf90(fixed = phe_X ~ 1,
random = ~ gg,
genetic = list(model = 'add_animal',
pedigree = globulus[, 1:3],
id = 'self'),
data = globulus)
Animal model: results
-
gg explains almost the same amount of phenotypic variability
-
The (additive) genetic effect explains part of the formerly residual variance
-
The heritability is computed automatically as $$h^2 = \frac{\sigma_a^2}{\sigma_a^2 + \sigma_{gg}^2+ \sigma^2}$$
summary(res.animal)
Extracting Predicted Breeding Values
## Predicted Breeding Values
# for the full pedigree first, and for the observed individuals
# by matrix multiplication with the incidence matrix
PBV.full <- ranef(res.animal)$genetic
PBV <- model.matrix(res.animal)$genetic %*% PBV.full
# Predicted genetic values vs.
# phenotype.
# Note: fitted = mu + PBV
qplot(fitted(res.animal), phe_X,
data = globulus) +
geom_abline(intercept = 0,
slope = 1,
col = 'gray')
Handling pedigrees
-
The pedigree needs to meet certain conditions
-
If it does not, breedR automatically completes, recodes and sorts
-
If recoding is necessary, breedR issues a warning because you need to be careful when retrieving results
-
See this guide for more details
Spatial autocorrelation

What is spatial autocorrelation
-
The residuals of any LMM must be noise
-
However, most times there are environmental factors that affect the response
-
This causes that observations that are close to each other tend to be more similar that observations that are far away
-
This is called spatial autocorrelation
-
It may affect both the estimations and their accuracy
-
This is why experiments are randomized into spatial blocks
Diagnosing spatial autocorrelation | residuals spatial plot
-
You can plot() the spatial arrangement of various model components (e.g. residuals)
-
Look like independent gaussian observations (i.e. noise)?
-
Do you see any signal in the background?
## Since coordinates have not
## been passed before they
## must be provided explicitly.
coordinates(res.animal) <-
globulus[, c('x', 'y')]
plot(res.animal, 'resid')
Diagnosing spatial autocorrelation | variograms of residuals
- Plot the variogram of residuals with
variogram()
variogram(res.animal)
Interpreting the variograms
- Isotropic variogram:
$$
\gamma(h) = \frac12 V[Z(\mathbf{u}) - Z(\mathbf{v})], \quad \text{dist}(\mathbf{u}, \mathbf{v}) = h
$$
The empirical isotropic variogram is built by aggregating
all the pairs of points separated by $h$, no matter the direction.
variogram(res.animal,
coord = globulus[, c('x', 'y')],
plot = 'isotropic')
ang <- seq(0, 2*pi, by = pi/6)[- (1+3*(0:4))]
# Colours from
# http://www.cookbook-r.com/Graphs/Colors_(ggplot2)/#a-colorblind-friendly-palette
cbPalette <- c("#E69F00", "#56B4E9", "#009E73", "#F0E442")
coord <- data.frame(x = cos(ang),
y = sin(ang))
ggplot(coord, aes(x, y)) +
geom_point(size = 6, col = cbPalette[1]) +
geom_point(aes(x = 0, y = 0), size = 6) +
coord_fixed() +
scale_x_continuous(breaks=NULL) +
scale_y_continuous(breaks=NULL) +
theme_minimal() +
theme(axis.title.x = element_blank(),
axis.title.y = element_blank())
Interpreting the variograms
- Row/Column variogram:
$$
\gamma(x, y) = \frac12 V[Z(\mathbf{u}) - Z(\mathbf{v})], \quad \text{dist}(\mathbf{u}, \mathbf{v}) = (x, y)
$$
The empirical row/col variogram is built by aggregating
all the pairs of points separated by exactly $x$ rows and $y$ columns.
variogram(res.animal,
coord = globulus[, c('x', 'y')],
plot = 'heat')
ggplot(coord, aes(x, y)) +
geom_point(size = 6, col = cbPalette[rep(c(1, 2, 2, 1), 2)]) +
geom_point(aes(x = 0, y = 0), size = 6) +
coord_fixed() +
scale_x_continuous(breaks=NULL) +
scale_y_continuous(breaks=NULL) +
theme_minimal() +
theme(axis.title.x = element_blank(),
axis.title.y = element_blank())
Interpreting the variograms
- Anisotropic variogram:
$$
\gamma(\mathbf{x}) = \frac12 V[Z(\mathbf{u}) - Z(\mathbf{v})], \quad \mathbf{u} = \mathbf{v} \pm \mathbf{x}
$$
The empirical anisotropic variogram is built by aggregating
all the pairs of points in the same direction separated by $|\mathbf{x}|$.
variogram(res.animal,
coord = globulus[, c('x', 'y')],
plot = 'aniso')
ggplot(coord, aes(x, y)) +
geom_point(size = 6, col = cbPalette[rep(1:4, 2)]) +
geom_point(aes(x = 0, y = 0), size = 6) +
coord_fixed() +
scale_x_continuous(breaks=NULL) +
scale_y_continuous(breaks=NULL) +
theme_minimal() +
theme(axis.title.x = element_blank(),
axis.title.y = element_blank())
Accounting for spatial autocorrelation
-
Include an explicit spatial effect in the model
-
I.e., a random effect with a specific covariance structure that reflects the spatial relationship between individuals
-
The block effect, is a very particular case:
- It is designed from the begining, possibly using prior knowledge
- Introduces independent effects between blocks
- Most neighbours are within the same block (i.e. share the same effect)
The blocks model
# The genetic component (DRY)
gen.globulus <- list(model = 'add_animal',
pedigree = globulus[, 1:3],
id = 'self')
res.blk <- remlf90(fixed = phe_X ~ 1,
random = ~ gg,
genetic = gen.globulus,
spatial = list(model = 'blocks',
coord = globulus[, c('x', 'y')],
id = 'bl'),
data = globulus)
- The
blocks spatial model is equivalent to random = ~ bl, but:
- specifying
coord is convenient for plotting (remember?)
blocks behaves as expected, even if bl is not a factor
Animal-spatial model: results
summary(res.blk)
- Now the additive-genetic variance and the heritability have increased! ($3.6$ and $0.18$ before)
Variogram of residuals
variogram(res.blk)
- There seems to remain some intra-block spatial autocorrelation
B-Splines model
- A continuous and smooth spatial surface built from a linear combination of basis functions
- The coefficients are modelled as a random effect
## Use the `em` method! `ai` does not like splines
res.spl <- remlf90(fixed = phe_X ~ 1,
random = ~ gg,
genetic = gen.globulus,
spatial = list(model = 'splines',
coord = globulus[, c('x','y')]),
data = globulus, method = 'em')
Autoregressive model
- A separable kronecker product of First order Autoregressive processes on the rows and the colums
res.ar1 <- remlf90(fixed = phe_X ~ 1,
random = ~ gg,
genetic = gen.globulus,
spatial = list(model = 'AR',
coord = globulus[, c('x','y')]),
data = globulus)
Change in model residuals
- We preserve the scale by using
compare.plots()
compare.plots(
list(`Animal model only` = plot(res.animal, 'residuals'),
`Animal/blocks model` = plot(res.blk, 'residuals'),
`Animal/splines model` = plot(res.spl, 'residuals'),
`Animal/AR1 model` = plot(res.ar1, 'residuals')))
Comparison of spatial components
compare.plots(list(Blocks = plot(res.blk, type = 'spatial'),
Splines = plot(res.spl, type = 'spatial'),
AR1xAR1 = plot(res.ar1, type = 'spatial')))
Prediction of the spatial effect in unobserved locations
-
The type fullspatial fills the holes (when possible)
-
See ?plot.remlf90
compare.plots(list(Blocks = plot(res.blk, type = 'fullspatial'),
Splines = plot(res.spl, type = 'fullspatial'),
AR1xAR1 = plot(res.ar1, type = 'fullspatial')))
Spatial parameters | Number of knots of a splines model
-
The smoothness of the spatial surface can be controlled modifying the number of base functions
-
This is, directly determined by the number of knots (nok) in each dimension
-
n.knots can be used as an additional argument in the spatial effect as a numeric vector of size 2.
-
Otherwise, is determined by the function given in breedR.getOption('splines.nok')
ggplot(transform(data.frame(x = 10:1e3),
nok = breedR:::determine.n.knots(x)),
aes(x, nok)) +
geom_line()
Spatial parameters | Autoregressive parameters of a AR model
-
Analogously, the patchiness of the AR effects can be controlled by the autoregressive parameter for each dimension
-
rho can be given as an additional argument in the spatial effect as a numeric vector of size 2
-
By default, breedR runs all the combinations in the grid produced by the values from breedR.getOption('ar.eval') and returns the one with largest likelihood
-
It returns also the full table of combinations and likelihoods in res$rho
Exercise | Tuning spatial parameters
-
Tuning parameters:
- model
splines: n.knots
- model
AR: rho
-
Increase the number of knots in the splines model and see if it improves the fit
-
Visualize the log-likelihood of the fitted AR models
-
Refine the grid around the most likely values, and refit using rho = rho.grid, where
rho.grid <- expand.grid(rho_r = seq(.7, .95, length = 4),
rho_c = seq(.7, .95, length = 4))
- What are now the most likely parameters?
Spatial #1 | B-splines model with increased nok
nok were (6, 6) by default (see summary())
res.spl99 <- remlf90(fixed = phe_X ~ 1, random = ~ gg,
genetic = gen.globulus,
spatial = list(model = 'splines',
coord = globulus[, c('x','y')],
n.knots = c(9, 9)),
data = globulus, method = 'em')
summary(res.spl)
summary(res.spl99)
Spatial #2 | Visualize log-likelihoods
qplot(rho_r, rho_c,
fill = loglik,
geom = 'tile',
data = res.ar1$rho)
knitr::kable(res.ar1$rho)
Spatial #3 | Refine grid
rho.grid <- expand.grid(rho_r = seq(.7, .95, length = 4),
rho_c = seq(.7, .95, length = 4))
res.ar.grid <- remlf90(fixed = phe_X ~ gg,
genetic = list(model = 'add_animal',
pedigree = globulus[,1:3],
id = 'self'),
spatial = list(model = 'AR',
coord = globulus[, c('x','y')],
rho = rho.grid),
data = globulus)
summary(res.ar.grid)
Competition
Theoretical model

-
Each individual have two (unknown) Breeding Values (BV)
-
The direct BV affects its own phenotype, while the competition BV affects its neghbours' (as the King moves)
-
The effect of the neighbouring competition BVs is given by their sum weighted by $1/d^\alpha$, where $\alpha$ is a tuning parameter called decay
-
Each set of BVs is modelled as a zero-mean random effect with structure matrix given by the pedigree and independent variances $\sigma^2_a$ and $\sigma^2_c$
-
Both random effects are modelled jointly with correlation $\rho$
Permanent Environmental Effect (pec)
-
Optional effect with environmental (rather than genetic) basis
-
Modelled as an individual independent random effect that affects neighbouring trees in the same (weighted) way
Simulation of data
breedR implements a convenient dataset simulator which keeps a similar syntax.
- See
?simulation for details on the syntax
# Simulation parameters
grid.size <- c(x=20, y=25) # cols/rows
coord <- expand.grid(sapply(grid.size,
seq))
Nobs <- prod(grid.size)
Nparents <- c(mum = 20, dad = 20)
sigma2_a <- 2 # direct add-gen var
sigma2_c <- 1 # compet add-gen var
rho <- -.7 # gen corr dire-comp
sigma2_s <- 1 # spatial variance
sigma2_p <- .5 # pec variance
sigma2 <- .5 # residual variance
S <- matrix(c(sigma2_a,
rho*sqrt(sigma2_a*sigma2_c),
rho*sqrt(sigma2_a*sigma2_c),
sigma2_c),
2, 2)
set.seed(12345)
simdat <-
breedR.sample.phenotype(
fixed = c(beta = 10),
genetic = list(model = 'competition',
Nparents = Nparents,
sigma2_a = S,
check.factorial=FALSE,
pec = sigma2_p),
spatial = list(model = 'AR',
grid.size = grid.size,
rho = c(.3, .8),
sigma2_s = sigma2_s),
residual.variance = sigma2
)
## Remove founders
dat <- subset(simdat,
!(is.na(simdat$sire)
& is.na(simdat$dam)))
Fitting a competition model
system.time(
res.comp <- remlf90(fixed = phenotype ~ 1,
genetic = list(model = 'competition',
pedigree = dat[, 1:3],
id = 'self',
coord = dat[, c('x', 'y')],
competition_decay = 1,
pec = list(present = TRUE)),
spatial = list(model = 'AR',
coord = dat[, c('x', 'y')],
rho = c(.3, .8)),
data = dat,
method = 'em') # AI diverges
)
True vs. estimated parameters
var.comp <-
data.frame(True = c(sigma2_a, sigma2_c, rho, sigma2_s, sigma2_p, sigma2),
Estimated = with(res.comp$var,
round(c(genetic[1, 1], genetic[2, 2],
genetic[1, 2]/sqrt(prod(diag(genetic))),
spatial, pec, Residual), digits = 2)),
row.names = c('direct', 'compet.', 'correl.',
'spatial', 'pec', 'residual'))
knitr::kable(var.comp)
labels <- c(paste0(rep('sigma', 5),
c('[A]', '[C]', '[S]', '[P]', ''), '^2'),
'rho')[c(1:2, 6, 3:5)]
ggplot(var.comp, aes(True, Estimated)) +
geom_point() +
geom_abline(intercept = 0, slope = 1, col = 'darkgray') +
geom_text(label = labels, parse = TRUE, hjust = -.5) +
expand_limits(x = 2.4, y = 2.6)
Exercise | Competition models
-
Plot the true vs predicted:
- direct and competition Breeding Values
- spatial effects
- pec effects
-
Plot the residuals and their variogram
- Do you think the residuals are independent?
- How would you improve the analysis?
Competition #1 | True vs. predicted components
## compute the predicted effects for the observations
## by matrix multiplication of the incidence matrix and the BLUPs
pred <- list()
Zd <- model.matrix(res.comp)$'genetic_direct'
pred$direct <- Zd %*% ranef(res.comp)$'genetic_direct'
## Watch out! for the competition effects you need to use the incidence
## matrix of the direct genetic effect, to get their own value.
## Otherwise, you get the predicted effect of the neighbours on each
## individual.
pred$comp <- Zd %*% ranef(res.comp)$'genetic_competition'
pred$pec <- model.matrix(res.comp)$pec %*% ranef(res.comp)$pec
Competition #1 | True vs. predicted components
comp.pred <-
rbind(
data.frame(
Component = 'direct BV',
True = dat$BV1,
Predicted = pred$direct),
data.frame(
Component = 'competition BV',
True = dat$BV2,
Predicted = pred$comp),
data.frame(
Component = 'pec',
True = dat$pec,
Predicted = as.vector(pred$pec)))
ggplot(comp.pred,
aes(True, Predicted)) +
geom_point() +
geom_abline(intercept = 0, slope = 1,
col = 'darkgray') +
facet_grid(~ Component)
The predition of the Permanent Environmental Competition effect is not precisely great...
Competition #2 | Map of residuals and their variogram
plot(res.comp, type = 'resid')
variogram(res.comp)
Generic component
The Generic model
This additional component allows to introduce a random effect $\psi$ with
arbitrary incidence and covariance matrices $Z$ and $\Sigma$:
$$
\begin{aligned}
y = & \mu + X \beta + Z \psi + \varepsilon \
\psi \sim & N(0, \sigma_\psi^2 \Sigma_\psi) \
\varepsilon \sim & N(0, \sigma_\varepsilon^2)
\end{aligned}
$$
Implementation of the generic component
## Fit a blocks effect using generic
inc.mat <- model.matrix(~ 0 + bl, globulus)
cov.mat <- diag(nlevels(globulus$bl))
res.blg <- remlf90(fixed = phe_X ~ gg,
generic = list(block = list(inc.mat,
cov.mat)),
data = globulus)
Example of result
summary(res.blg)
Prediction
Predicting values for unobserved trees
-
You can predict the Breeding Value of an unmeasured tree
-
Or the expected phenotype of a death tree (or an hypothetical scenario)
-
Information is gathered from the covariates, the spatial structure and the pedigree
-
Simply include the individual in the dataset with the response set as NA
Leave-one-out cross-validation
-
Re-fit the simulated competition data with one measurement removed
-
Afterwards, compare the predicted values for the unmeasured individuals with their true simulated values
rm.idx <- 8
rm.exp <- with(dat[rm.idx, ],
phenotype - resid)
dat.loo <- dat
dat.loo[rm.idx, 'phenotype'] <- NA
res.comp.loo <- remlf90(fixed = phenotype ~ 1,
genetic = list(model = 'competition',
pedigree = dat[, 1:3],
id = 'self',
coord = dat[, c('x', 'y')],
competition_decay = 1,
pec = list(present = TRUE)),
spatial = list(model = 'AR',
coord = dat[, c('x', 'y')],
rho = c(.3, .8)),
data = dat.loo,
method = 'em')
## compute the predicted effects for the observations
## by matrix multiplication of the incidence matrix and the BLUPs
Zd <- model.matrix(res.comp)$'genetic_direct'
pred.BV.loo.mat <- with(ranef(res.comp.loo),
cbind(`genetic_direct`, `genetic_competition`))
pred.genetic.loo <- Zd[rm.idx, ] %*% pred.BV.loo.mat
valid.pred <-
data.frame(True = with(dat[rm.idx, ],
c(BV1, BV2, rm.exp)),
Pred.loo = c(pred.genetic.loo,
fitted(res.comp.loo)[rm.idx]),
row.names = c('direct BV', 'competition BV', 'exp. phenotype'))
knitr::kable(round(valid.pred, 2))
Exercise | Cross validation
-
Extend the last table to include the predicted values with the full dataset
-
Remove 1/10th of the phenotypes randomly, and predict their expected phenotype
- Have the parameter estimations changed too much?
-
Compute the Root Mean Square Error (RMSE) of Prediction with respect to the true values
Cross-validation #1 | Include prediction with full data
pred.BV.mat <- with(ranef(res.comp),
cbind(`genetic_direct`, `genetic_competition`))
valid.pred$Pred.full <- c(Zd[rm.idx, ] %*% pred.BV.mat,
fitted(res.comp)[rm.idx])
knitr::kable(round(valid.pred[,c(1,3,2)], 2))
Cross-validation #2 | Perform cross-validation on 1/10th of the observations
rm.idx <- sample(nrow(dat), nrow(dat)/10)
dat.cv <- dat
dat.cv[rm.idx, 'phenotype'] <- NA
## Re-fit the model and build table
res.comp.cv <-
remlf90(fixed = phenotype ~ 1,
genetic = list(model = 'competition',
pedigree = dat[, 1:3],
id = 'self',
coord = dat[, c('x', 'y')],
competition_decay = 1,
pec = list(present = TRUE)),
spatial = list(model = 'AR',
coord = dat[, c('x', 'y')],
rho = c(.3, .8)),
data = dat.cv,
method = 'em')
var.comp <-
data.frame(
'Fully estimated' = with(res.comp$var,
round(c(genetic[1, 1], genetic[2, 2],
genetic[1, 2]/sqrt(prod(diag(genetic))),
spatial, pec, Residual), digits = 2)),
'CV estimated' = with(res.comp.cv$var,
round(c(genetic[1, 1], genetic[2, 2],
genetic[1, 2]/sqrt(prod(diag(genetic))),
spatial, pec, Residual), digits = 2)),
row.names = c('direct', 'compet.', 'correl.',
'spatial', 'pec', 'residual')
)
knitr::kable(var.comp)
Cross-validation #3 | MSE of Prediction
true.exp.cv <- with(dat[rm.idx, ], phenotype - resid)
round(sqrt(mean((fitted(res.comp.cv)[rm.idx] - true.exp.cv)^2)), 2)
Multiple traits
breedR provides a basic interface for multi-trait models which only
requires specifying the different traits in the main formula using cbind().
## Filter site and select relevant variables
dat <-
droplevels(
douglas[douglas$site == "s3",
names(douglas)[!grepl("H0[^4]|AN|BR|site", names(douglas))]]
)
res <-
remlf90(
fixed = cbind(H04, C13) ~ orig,
genetic = list(
model = 'add_animal',
pedigree = dat[, 1:3],
id = 'self'),
data = dat
)
A full covariance matrix across traits is estimated for each random effect,
and all results, including heritabilities, are expressed effect-wise:
summary(res)
Although the results are summarized in tabular form, the covariance matrices
can be recovered directly:
res$var[["genetic", "Estimated variances"]]
## Use cov2cor() to compute correlations
cov2cor(res$var[["genetic", "Estimated variances"]])
Estimates of fixed effects and BLUPs of random effects can be recovered with
fixef() and ranef() as usual. The only difference is that they will return a
list of matrices rather than vectors, with one column per trait.
The standard errors are given as attributes, and are displayed in tabular form
whenever the object is printed.
fixef(res) ## printed in tabular form, but...
unclass(fixef(res)) ## actually a matrix of estimates with attribute "se"
str(ranef(res))
head(ranef(res)$genetic)
Recovering the breeding values for each observation in the original dataset
follows the same procedure as for one trait: multiply the incidence matrix by
the BLUP matrix. The result, however, will be a matrix with one column per
trait.
head(model.matrix(res)$genetic %*% ranef(res)$genetic)
Initial (co)variance specification
breedR will use the empirical variances and covariances to compute initial
covariance matrices. But you can specify your own. This is particularly
interesting for setting some initial covariances to 0, which indicates that you
don't want that component to be estimated, and thus reducing the dimension of
the model.
Typical cases are Multi-Environment Trials (MET, e.g. multiple sites,
or years) where you don't really want to estimate the residual covariances, or
when you know a priori that two traits are little correlated.
Specify the initial covariance values in matrix form.
initial_covs <- list(
genetic = 1e3*matrix(c(1, .5, .5, 1), nrow = 2),
residual = diag(2) # no residual covariances
)
res <-
remlf90(
fixed = cbind(H04, C13) ~ orig,
genetic = list(
model = 'add_animal',
pedigree = dat[, 1:3],
id = 'self',
var.ini = initial_covs$genetic),
data = dat,
var.ini = list(residual = initial_covs$residual)
)
Some more features
Metagene interface
-
We have used simulated data from the metagene software
-
If you simulate data, import the results with read.metagene()
-
Use several common methods with a metagene object:
summary(), plot(), as.data.frame()
-
Plus some more specific metagene functions:
b.values(), get.ntraits(), ngenerations(), nindividuals(), get.pedigree()
-
And specific functions about spatial arrangement:
coordinates() extract coordinatessim.spatial() simulates some spatial autocorrelation
Simulation framework
-
The function breedR.sample.phenotype() simulates datasets from all the model structures available in breedR
-
Limitation: only one generation, with random matings of founders
-
See ?simulation for details
Remote computation
If you have access to a Linux server through SSH, you can perform computations remotely
-
Take advantage of more memory or faster processors
-
Parallelize jobs
-
Free local resources while fitting models
-
See ?remote for details
Package options
-
breedR features a list of configurable options
-
Use breedR.setOption(...) for changing an option during the current sesion
-
Set options permanently in the file $HOME/.breedRrc
-
see ?breedR.option for details
breedR.getOption()
famuvie/breedR documentation built on Aug. 6, 2024, 9:10 p.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
library(knitr) opts_chunk$set(echo = TRUE, message = TRUE, warning = TRUE, # comment = NA, fig.width = 4, fig.height = 3, cache = TRUE, autodep = TRUE, ## invalidate cache when either versions of R or breedR change ## http://yihui.name/knitr/demo/cache/ cache.extra= list(R.version, packageVersion('breedR')))
library(breedR) library(ggplot2)
Intro
What is breedR
- R-package implementing statistical models specifically suited for forest genetic resources analysts.
- Ultimately Mixed Models, but not necessarily easy to implement and use
-
breedR acts as an interface which provides the means to:
- Combine any number of these models as components of a larger model
- Compute automatically incidence and covariance matrices from a few input parameters
- Fit the model
- Plot data and results, and perform model diagnostics
Installation
-
Project web page http://famuvie.github.io/breedR/
- Set up this URL as a package repository in
.Rprofile(detailed instructions on the web) install.packages('breedR')- Not possible to use
CRANdue to closed-sourceBLUPF90programs
- Set up this URL as a package repository in
-
GitHub dev-site https://github.com/famuvie/breedR
if( !require(devtools) ) install.packages('devtools')devtools::install_github('famuvie/breedR')
Where to find help
-
Package's help:
help(package = breedR)- Help pages
?remlf90 - Code demos
demo(topic, package = 'breedR')(omittopicfor a list) - Vignettes
vignette(package = 'breedR')(pkg and wiki)
- Help pages
-
- Guides, tutorials, FAQ
-
Mailing list http://groups.google.com/group/breedr
- Questions and debates about usage and interface
-
- Bug reports
- Feature requests
License
![]()
- breedR is FOSS. Licensed GPL-3
RShowDoc('LICENSE', package = 'breedR')
- You can use and distribute breedR for any purpose
-
You can modify it to suit your needs
- we encourage to!
- please consider contributing your improvements
- you can distribute your modified version under the GPL
-
However, breedR makes (intensive) use of the
BLUPF90suite of Fortran programs, which are for free but not free (remember CRAN?)
Roadmap | Future developments
-
Bayesian inference
-
Multi-trait support
-
Genotype$\times$Environment interaction
-
Support for longitudinal data
Functionality
Inference
Frequentist
-
Currently, only frequentist inference is supported via REML estimation of variance components.
-
The function
remlf90(), provides an interface to bothREMLF90andAIREMLF90functions in theBLUPF90suite of Fortran programs. -
Type
?remlf90for details on the syntax
Bayesian
-
It's on the roadmap for the next year
-
Will use a gibbs sampler from
BLUPF90, and possibly alsoINLA -
The interface will change a bit, separating the model specification from the fit
Linear Mixed Models with unstructured random effects
Example dataset
knitr::kable(head(globulus))
cat(attr(globulus, 'comment'), sep ='\n') str(globulus, give.attr = FALSE)
A simple Provenance Test
Specify the genetic group gg as an unstructured random effect using the standard formulas in R
$$ \begin{aligned} \mathrm{phe}X = & \mu + Z \mathrm{gg} + \varepsilon \ \mathrm{gg} \sim & N(0, \sigma{\mathrm{gg}}^2) \ \varepsilon \sim & N(0, \sigma_\varepsilon^2) \end{aligned} $$
res <- remlf90(fixed = phe_X ~ 1, random = ~ gg, data = globulus)
Initial variances specification
To avoid the notification, initial values for all the variance components must be made explicit using the argument var.ini:
res <- remlf90(fixed = phe_X ~ 1, random = ~ gg, var.ini = list(gg = 2, resid = 10), data = globulus)
Although in most cases the results will not change at all, we encourage to give explicit initial values for variance components. Specially when some estimate can be artifact. This is also useful for checking sensitivity to initial values.
Exploring the results
summary(res)
- Note that
AI-REMLhas been used by default. - You can also specify
method = 'em'. - Learn about the difference.
Further extractor functions
fixef(res) ranef(res)
Further extractor functions
qplot( fitted(res), globulus$phe_X) + geom_abline(intercept = 0, slope = 1, col = 'darkgrey') str(resid(res)) extractAIC(res) logLik(res)
Hierarchical and Factorial models
-
In globulus, the family (
mum) is nested within the provenance (gg) -
This is a matter of codification:
Nested factors
|gg | mum | |---:|:----| |A | 1 | |A | 2 | |B | 3 | |B | 4 |
Crossed factors
|gg | mum | |---:|:----| |A | 1 | |A | 2 | |B | 1 | |B | 2 |
Model specification
- Otherwise, in both cases we specify the model in the same way:
random = ~ gg + factor(mum) # note that mum is numeric
- Furthermore, this approach can handle unbalanced and mixed designs
Interactions
- Standard
Rnotation:
random = ~ gg * factor(mum)
-
Not available yet (feature request?)
-
Workaround: build the interaction variable manually
-
Example:
ggandblockare crossed factors
dat <- transform(globulus, interaction = factor(gg:bl)) random = ~ gg + bl + interaction
Exercise | Hierarchical and Factorial models
-
Use
remlf90()and the globulus dataset to fit- a hierarchical model using
mumwithingg - a factorial model using
ggandbl
- a hierarchical model using
-
Explore the results with
summary()- is the family (
mum) effect relevant? - is there any evidence of interaction between
ggandbl?
- is the family (
Hierarchical and Factorial models #1 | Fitting models
res.h <- remlf90(fixed = phe_X ~ 1, random = ~ factor(mum) + gg, data = globulus)
# Interaction variable globulus.f <- transform(globulus, gg_bl = factor(gg:bl)) res.f <- remlf90(fixed = phe_X ~ 1, random = ~ gg + bl + gg_bl, data = globulus.f)
Hierarchical and Factorial models #2 | Hierarchical model
- The family effect is not very important, in terms of explained variance
- However, the model is a bit better with it (AIC, logLik)
summary(res) summary(res.h)
Hierarchical and Factorial models #3 | Factorial model
- Looks like the interaction between block and provenance is negligible
- (apart from the fact that it makes no sense at all, and shuld not have been even considered in the first place)
- compare with the model without interaction
summary(res.f)
## result without interaction res.f0 <- remlf90(fixed = phe_X ~ 1, random = ~ gg + bl, data = globulus) paste('AIC:', round(extractAIC(res.f0)), 'logLik:', round(logLik(res.f0)))
Additive Genetic Effect
What is an additive genetic effect
Random effect at individual level
Based on a pedigree
BLUP of Breeding Values from own and relatives' phenotypes
Represents the additive component of the genetic value
More general:
- family effect is a particular case
- accounts for more than one generation
- mixed relationships
More flexible: allows to select individuals within families
Specifying a pedigree
-
A 3-column
data.frameormatrixwith the codes for each individual and its parents -
A family effect is easily translated into a pedigree:
- use the family code as the identification of a fictitious mother
- use
0orNAas codes for the unknown fathers
knitr::kable(head(globulus[, 1:3]))
Fitting an animal model
res.animal <- remlf90(fixed = phe_X ~ 1, random = ~ gg, genetic = list(model = 'add_animal', pedigree = globulus[, 1:3], id = 'self'), data = globulus)
Animal model: results
-
ggexplains almost the same amount of phenotypic variability -
The (additive)
geneticeffect explains part of the formerly residual variance -
The heritability is computed automatically as $$h^2 = \frac{\sigma_a^2}{\sigma_a^2 + \sigma_{gg}^2+ \sigma^2}$$
summary(res.animal)
Extracting Predicted Breeding Values
## Predicted Breeding Values # for the full pedigree first, and for the observed individuals # by matrix multiplication with the incidence matrix PBV.full <- ranef(res.animal)$genetic PBV <- model.matrix(res.animal)$genetic %*% PBV.full # Predicted genetic values vs. # phenotype. # Note: fitted = mu + PBV qplot(fitted(res.animal), phe_X, data = globulus) + geom_abline(intercept = 0, slope = 1, col = 'gray')
Handling pedigrees
-
The pedigree needs to meet certain conditions
-
If it does not, breedR automatically completes, recodes and sorts
-
If recoding is necessary, breedR issues a warning because you need to be careful when retrieving results
-
See this guide for more details
Spatial autocorrelation
What is spatial autocorrelation
-
The residuals of any LMM must be noise
-
However, most times there are environmental factors that affect the response
-
This causes that observations that are close to each other tend to be more similar that observations that are far away
-
This is called spatial autocorrelation
-
It may affect both the estimations and their accuracy
-
This is why experiments are randomized into spatial blocks
Diagnosing spatial autocorrelation | residuals spatial plot
-
You can
plot()the spatial arrangement of various model components (e.g. residuals) -
Look like independent gaussian observations (i.e. noise)?
-
Do you see any signal in the background?
## Since coordinates have not ## been passed before they ## must be provided explicitly. coordinates(res.animal) <- globulus[, c('x', 'y')] plot(res.animal, 'resid')
Diagnosing spatial autocorrelation | variograms of residuals
- Plot the variogram of residuals with
variogram()
variogram(res.animal)
Interpreting the variograms
- Isotropic variogram: $$ \gamma(h) = \frac12 V[Z(\mathbf{u}) - Z(\mathbf{v})], \quad \text{dist}(\mathbf{u}, \mathbf{v}) = h $$
The empirical isotropic variogram is built by aggregating all the pairs of points separated by $h$, no matter the direction.
variogram(res.animal, coord = globulus[, c('x', 'y')], plot = 'isotropic')
ang <- seq(0, 2*pi, by = pi/6)[- (1+3*(0:4))] # Colours from # http://www.cookbook-r.com/Graphs/Colors_(ggplot2)/#a-colorblind-friendly-palette cbPalette <- c("#E69F00", "#56B4E9", "#009E73", "#F0E442") coord <- data.frame(x = cos(ang), y = sin(ang)) ggplot(coord, aes(x, y)) + geom_point(size = 6, col = cbPalette[1]) + geom_point(aes(x = 0, y = 0), size = 6) + coord_fixed() + scale_x_continuous(breaks=NULL) + scale_y_continuous(breaks=NULL) + theme_minimal() + theme(axis.title.x = element_blank(), axis.title.y = element_blank())
Interpreting the variograms
- Row/Column variogram: $$ \gamma(x, y) = \frac12 V[Z(\mathbf{u}) - Z(\mathbf{v})], \quad \text{dist}(\mathbf{u}, \mathbf{v}) = (x, y) $$
The empirical row/col variogram is built by aggregating all the pairs of points separated by exactly $x$ rows and $y$ columns.
variogram(res.animal, coord = globulus[, c('x', 'y')], plot = 'heat')
ggplot(coord, aes(x, y)) + geom_point(size = 6, col = cbPalette[rep(c(1, 2, 2, 1), 2)]) + geom_point(aes(x = 0, y = 0), size = 6) + coord_fixed() + scale_x_continuous(breaks=NULL) + scale_y_continuous(breaks=NULL) + theme_minimal() + theme(axis.title.x = element_blank(), axis.title.y = element_blank())
Interpreting the variograms
- Anisotropic variogram: $$ \gamma(\mathbf{x}) = \frac12 V[Z(\mathbf{u}) - Z(\mathbf{v})], \quad \mathbf{u} = \mathbf{v} \pm \mathbf{x} $$
The empirical anisotropic variogram is built by aggregating all the pairs of points in the same direction separated by $|\mathbf{x}|$.
variogram(res.animal, coord = globulus[, c('x', 'y')], plot = 'aniso')
ggplot(coord, aes(x, y)) + geom_point(size = 6, col = cbPalette[rep(1:4, 2)]) + geom_point(aes(x = 0, y = 0), size = 6) + coord_fixed() + scale_x_continuous(breaks=NULL) + scale_y_continuous(breaks=NULL) + theme_minimal() + theme(axis.title.x = element_blank(), axis.title.y = element_blank())
Accounting for spatial autocorrelation
-
Include an explicit spatial effect in the model
-
I.e., a random effect with a specific covariance structure that reflects the spatial relationship between individuals
-
The block effect, is a very particular case:
- It is designed from the begining, possibly using prior knowledge
- Introduces independent effects between blocks
- Most neighbours are within the same block (i.e. share the same effect)
The blocks model
# The genetic component (DRY) gen.globulus <- list(model = 'add_animal', pedigree = globulus[, 1:3], id = 'self') res.blk <- remlf90(fixed = phe_X ~ 1, random = ~ gg, genetic = gen.globulus, spatial = list(model = 'blocks', coord = globulus[, c('x', 'y')], id = 'bl'), data = globulus)
- The
blocksspatial model is equivalent torandom = ~ bl, but:- specifying
coordis convenient for plotting (remember?) blocksbehaves as expected, even ifblis not a factor
- specifying
Animal-spatial model: results
summary(res.blk)
- Now the additive-genetic variance and the heritability have increased! ($3.6$ and $0.18$ before)
Variogram of residuals
variogram(res.blk)
- There seems to remain some intra-block spatial autocorrelation
B-Splines model
- A continuous and smooth spatial surface built from a linear combination of basis functions
- The coefficients are modelled as a random effect
## Use the `em` method! `ai` does not like splines res.spl <- remlf90(fixed = phe_X ~ 1, random = ~ gg, genetic = gen.globulus, spatial = list(model = 'splines', coord = globulus[, c('x','y')]), data = globulus, method = 'em')
Autoregressive model
- A separable kronecker product of First order Autoregressive processes on the rows and the colums
res.ar1 <- remlf90(fixed = phe_X ~ 1, random = ~ gg, genetic = gen.globulus, spatial = list(model = 'AR', coord = globulus[, c('x','y')]), data = globulus)
Change in model residuals
- We preserve the scale by using
compare.plots()
compare.plots( list(`Animal model only` = plot(res.animal, 'residuals'), `Animal/blocks model` = plot(res.blk, 'residuals'), `Animal/splines model` = plot(res.spl, 'residuals'), `Animal/AR1 model` = plot(res.ar1, 'residuals')))
Comparison of spatial components
compare.plots(list(Blocks = plot(res.blk, type = 'spatial'), Splines = plot(res.spl, type = 'spatial'), AR1xAR1 = plot(res.ar1, type = 'spatial')))
Prediction of the spatial effect in unobserved locations
-
The type
fullspatialfills the holes (when possible) -
See
?plot.remlf90
compare.plots(list(Blocks = plot(res.blk, type = 'fullspatial'), Splines = plot(res.spl, type = 'fullspatial'), AR1xAR1 = plot(res.ar1, type = 'fullspatial')))
Spatial parameters | Number of knots of a splines model
-
The smoothness of the spatial surface can be controlled modifying the number of base functions
-
This is, directly determined by the number of knots (nok) in each dimension
-
n.knotscan be used as an additional argument in thespatialeffect as a numeric vector of size 2. -
Otherwise, is determined by the function given in
breedR.getOption('splines.nok')
ggplot(transform(data.frame(x = 10:1e3), nok = breedR:::determine.n.knots(x)), aes(x, nok)) + geom_line()
Spatial parameters | Autoregressive parameters of a AR model
-
Analogously, the patchiness of the
AReffects can be controlled by the autoregressive parameter for each dimension -
rhocan be given as an additional argument in thespatialeffect as a numeric vector of size 2 -
By default, breedR runs all the combinations in the grid produced by the values from
breedR.getOption('ar.eval')and returns the one with largest likelihood -
It returns also the full table of combinations and likelihoods in
res$rho
Exercise | Tuning spatial parameters
-
Tuning parameters:
- model
splines:n.knots - model
AR:rho
- model
-
Increase the number of knots in the
splinesmodel and see if it improves the fit -
Visualize the log-likelihood of the fitted
ARmodels -
Refine the grid around the most likely values, and refit using
rho = rho.grid, where
rho.grid <- expand.grid(rho_r = seq(.7, .95, length = 4), rho_c = seq(.7, .95, length = 4))
- What are now the most likely parameters?
Spatial #1 | B-splines model with increased nok
nokwere (6, 6) by default (seesummary())
res.spl99 <- remlf90(fixed = phe_X ~ 1, random = ~ gg, genetic = gen.globulus, spatial = list(model = 'splines', coord = globulus[, c('x','y')], n.knots = c(9, 9)), data = globulus, method = 'em')
summary(res.spl) summary(res.spl99)
Spatial #2 | Visualize log-likelihoods
qplot(rho_r, rho_c, fill = loglik, geom = 'tile', data = res.ar1$rho)
knitr::kable(res.ar1$rho)
Spatial #3 | Refine grid
rho.grid <- expand.grid(rho_r = seq(.7, .95, length = 4), rho_c = seq(.7, .95, length = 4)) res.ar.grid <- remlf90(fixed = phe_X ~ gg, genetic = list(model = 'add_animal', pedigree = globulus[,1:3], id = 'self'), spatial = list(model = 'AR', coord = globulus[, c('x','y')], rho = rho.grid), data = globulus) summary(res.ar.grid)
Competition
Theoretical model
Each individual have two (unknown) Breeding Values (BV)
The
directBV affects its own phenotype, while thecompetitionBV affects its neghbours' (as the King moves)The effect of the neighbouring
competitionBVs is given by their sum weighted by $1/d^\alpha$, where $\alpha$ is a tuning parameter calleddecayEach set of BVs is modelled as a zero-mean random effect with structure matrix given by the pedigree and independent variances $\sigma^2_a$ and $\sigma^2_c$
Both random effects are modelled jointly with correlation $\rho$
Permanent Environmental Effect (pec)
-
Optional effect with environmental (rather than genetic) basis
-
Modelled as an individual independent random effect that affects neighbouring trees in the same (weighted) way
Simulation of data
breedR implements a convenient dataset simulator which keeps a similar syntax.
- See
?simulationfor details on the syntax
# Simulation parameters grid.size <- c(x=20, y=25) # cols/rows coord <- expand.grid(sapply(grid.size, seq)) Nobs <- prod(grid.size) Nparents <- c(mum = 20, dad = 20) sigma2_a <- 2 # direct add-gen var sigma2_c <- 1 # compet add-gen var rho <- -.7 # gen corr dire-comp sigma2_s <- 1 # spatial variance sigma2_p <- .5 # pec variance sigma2 <- .5 # residual variance S <- matrix(c(sigma2_a, rho*sqrt(sigma2_a*sigma2_c), rho*sqrt(sigma2_a*sigma2_c), sigma2_c), 2, 2) set.seed(12345) simdat <- breedR.sample.phenotype( fixed = c(beta = 10), genetic = list(model = 'competition', Nparents = Nparents, sigma2_a = S, check.factorial=FALSE, pec = sigma2_p), spatial = list(model = 'AR', grid.size = grid.size, rho = c(.3, .8), sigma2_s = sigma2_s), residual.variance = sigma2 ) ## Remove founders dat <- subset(simdat, !(is.na(simdat$sire) & is.na(simdat$dam)))
Fitting a competition model
system.time( res.comp <- remlf90(fixed = phenotype ~ 1, genetic = list(model = 'competition', pedigree = dat[, 1:3], id = 'self', coord = dat[, c('x', 'y')], competition_decay = 1, pec = list(present = TRUE)), spatial = list(model = 'AR', coord = dat[, c('x', 'y')], rho = c(.3, .8)), data = dat, method = 'em') # AI diverges )
True vs. estimated parameters
var.comp <- data.frame(True = c(sigma2_a, sigma2_c, rho, sigma2_s, sigma2_p, sigma2), Estimated = with(res.comp$var, round(c(genetic[1, 1], genetic[2, 2], genetic[1, 2]/sqrt(prod(diag(genetic))), spatial, pec, Residual), digits = 2)), row.names = c('direct', 'compet.', 'correl.', 'spatial', 'pec', 'residual')) knitr::kable(var.comp)
labels <- c(paste0(rep('sigma', 5), c('[A]', '[C]', '[S]', '[P]', ''), '^2'), 'rho')[c(1:2, 6, 3:5)] ggplot(var.comp, aes(True, Estimated)) + geom_point() + geom_abline(intercept = 0, slope = 1, col = 'darkgray') + geom_text(label = labels, parse = TRUE, hjust = -.5) + expand_limits(x = 2.4, y = 2.6)
Exercise | Competition models
-
Plot the true vs predicted:
- direct and competition Breeding Values
- spatial effects
- pec effects
-
Plot the residuals and their variogram
- Do you think the residuals are independent?
- How would you improve the analysis?
Competition #1 | True vs. predicted components
## compute the predicted effects for the observations ## by matrix multiplication of the incidence matrix and the BLUPs pred <- list() Zd <- model.matrix(res.comp)$'genetic_direct' pred$direct <- Zd %*% ranef(res.comp)$'genetic_direct' ## Watch out! for the competition effects you need to use the incidence ## matrix of the direct genetic effect, to get their own value. ## Otherwise, you get the predicted effect of the neighbours on each ## individual. pred$comp <- Zd %*% ranef(res.comp)$'genetic_competition' pred$pec <- model.matrix(res.comp)$pec %*% ranef(res.comp)$pec
Competition #1 | True vs. predicted components
comp.pred <- rbind( data.frame( Component = 'direct BV', True = dat$BV1, Predicted = pred$direct), data.frame( Component = 'competition BV', True = dat$BV2, Predicted = pred$comp), data.frame( Component = 'pec', True = dat$pec, Predicted = as.vector(pred$pec))) ggplot(comp.pred, aes(True, Predicted)) + geom_point() + geom_abline(intercept = 0, slope = 1, col = 'darkgray') + facet_grid(~ Component)
The predition of the Permanent Environmental Competition effect is not precisely great...
Competition #2 | Map of residuals and their variogram
plot(res.comp, type = 'resid') variogram(res.comp)
Generic component
The Generic model
This additional component allows to introduce a random effect $\psi$ with arbitrary incidence and covariance matrices $Z$ and $\Sigma$:
$$ \begin{aligned} y = & \mu + X \beta + Z \psi + \varepsilon \ \psi \sim & N(0, \sigma_\psi^2 \Sigma_\psi) \ \varepsilon \sim & N(0, \sigma_\varepsilon^2) \end{aligned} $$
Implementation of the generic component
## Fit a blocks effect using generic inc.mat <- model.matrix(~ 0 + bl, globulus) cov.mat <- diag(nlevels(globulus$bl)) res.blg <- remlf90(fixed = phe_X ~ gg, generic = list(block = list(inc.mat, cov.mat)), data = globulus)
Example of result
summary(res.blg)
Prediction
Predicting values for unobserved trees
-
You can predict the Breeding Value of an unmeasured tree
-
Or the expected phenotype of a death tree (or an hypothetical scenario)
-
Information is gathered from the covariates, the spatial structure and the pedigree
-
Simply include the individual in the dataset with the response set as
NA
Leave-one-out cross-validation
-
Re-fit the simulated competition data with one measurement removed
-
Afterwards, compare the predicted values for the unmeasured individuals with their true simulated values
rm.idx <- 8 rm.exp <- with(dat[rm.idx, ], phenotype - resid) dat.loo <- dat dat.loo[rm.idx, 'phenotype'] <- NA
res.comp.loo <- remlf90(fixed = phenotype ~ 1, genetic = list(model = 'competition', pedigree = dat[, 1:3], id = 'self', coord = dat[, c('x', 'y')], competition_decay = 1, pec = list(present = TRUE)), spatial = list(model = 'AR', coord = dat[, c('x', 'y')], rho = c(.3, .8)), data = dat.loo, method = 'em')
## compute the predicted effects for the observations ## by matrix multiplication of the incidence matrix and the BLUPs Zd <- model.matrix(res.comp)$'genetic_direct' pred.BV.loo.mat <- with(ranef(res.comp.loo), cbind(`genetic_direct`, `genetic_competition`)) pred.genetic.loo <- Zd[rm.idx, ] %*% pred.BV.loo.mat valid.pred <- data.frame(True = with(dat[rm.idx, ], c(BV1, BV2, rm.exp)), Pred.loo = c(pred.genetic.loo, fitted(res.comp.loo)[rm.idx]), row.names = c('direct BV', 'competition BV', 'exp. phenotype')) knitr::kable(round(valid.pred, 2))
Exercise | Cross validation
-
Extend the last table to include the predicted values with the full dataset
-
Remove 1/10th of the phenotypes randomly, and predict their expected phenotype
- Have the parameter estimations changed too much?
-
Compute the Root Mean Square Error (RMSE) of Prediction with respect to the true values
Cross-validation #1 | Include prediction with full data
pred.BV.mat <- with(ranef(res.comp), cbind(`genetic_direct`, `genetic_competition`)) valid.pred$Pred.full <- c(Zd[rm.idx, ] %*% pred.BV.mat, fitted(res.comp)[rm.idx]) knitr::kable(round(valid.pred[,c(1,3,2)], 2))
Cross-validation #2 | Perform cross-validation on 1/10th of the observations
rm.idx <- sample(nrow(dat), nrow(dat)/10) dat.cv <- dat dat.cv[rm.idx, 'phenotype'] <- NA ## Re-fit the model and build table res.comp.cv <- remlf90(fixed = phenotype ~ 1, genetic = list(model = 'competition', pedigree = dat[, 1:3], id = 'self', coord = dat[, c('x', 'y')], competition_decay = 1, pec = list(present = TRUE)), spatial = list(model = 'AR', coord = dat[, c('x', 'y')], rho = c(.3, .8)), data = dat.cv, method = 'em') var.comp <- data.frame( 'Fully estimated' = with(res.comp$var, round(c(genetic[1, 1], genetic[2, 2], genetic[1, 2]/sqrt(prod(diag(genetic))), spatial, pec, Residual), digits = 2)), 'CV estimated' = with(res.comp.cv$var, round(c(genetic[1, 1], genetic[2, 2], genetic[1, 2]/sqrt(prod(diag(genetic))), spatial, pec, Residual), digits = 2)), row.names = c('direct', 'compet.', 'correl.', 'spatial', 'pec', 'residual') ) knitr::kable(var.comp)
Cross-validation #3 | MSE of Prediction
true.exp.cv <- with(dat[rm.idx, ], phenotype - resid) round(sqrt(mean((fitted(res.comp.cv)[rm.idx] - true.exp.cv)^2)), 2)
Multiple traits
breedR provides a basic interface for multi-trait models which only
requires specifying the different traits in the main formula using cbind().
## Filter site and select relevant variables dat <- droplevels( douglas[douglas$site == "s3", names(douglas)[!grepl("H0[^4]|AN|BR|site", names(douglas))]] ) res <- remlf90( fixed = cbind(H04, C13) ~ orig, genetic = list( model = 'add_animal', pedigree = dat[, 1:3], id = 'self'), data = dat )
A full covariance matrix across traits is estimated for each random effect, and all results, including heritabilities, are expressed effect-wise:
summary(res)
Although the results are summarized in tabular form, the covariance matrices can be recovered directly:
res$var[["genetic", "Estimated variances"]] ## Use cov2cor() to compute correlations cov2cor(res$var[["genetic", "Estimated variances"]])
Estimates of fixed effects and BLUPs of random effects can be recovered with
fixef() and ranef() as usual. The only difference is that they will return a
list of matrices rather than vectors, with one column per trait.
The standard errors are given as attributes, and are displayed in tabular form whenever the object is printed.
fixef(res) ## printed in tabular form, but... unclass(fixef(res)) ## actually a matrix of estimates with attribute "se" str(ranef(res)) head(ranef(res)$genetic)
Recovering the breeding values for each observation in the original dataset follows the same procedure as for one trait: multiply the incidence matrix by the BLUP matrix. The result, however, will be a matrix with one column per trait.
head(model.matrix(res)$genetic %*% ranef(res)$genetic)
Initial (co)variance specification
breedR will use the empirical variances and covariances to compute initial covariance matrices. But you can specify your own. This is particularly interesting for setting some initial covariances to 0, which indicates that you don't want that component to be estimated, and thus reducing the dimension of the model.
Typical cases are Multi-Environment Trials (MET, e.g. multiple sites, or years) where you don't really want to estimate the residual covariances, or when you know a priori that two traits are little correlated.
Specify the initial covariance values in matrix form.
initial_covs <- list( genetic = 1e3*matrix(c(1, .5, .5, 1), nrow = 2), residual = diag(2) # no residual covariances ) res <- remlf90( fixed = cbind(H04, C13) ~ orig, genetic = list( model = 'add_animal', pedigree = dat[, 1:3], id = 'self', var.ini = initial_covs$genetic), data = dat, var.ini = list(residual = initial_covs$residual) )
Some more features
Metagene interface
-
We have used simulated data from the
metagenesoftware -
If you simulate data, import the results with
read.metagene() -
Use several common methods with a
metageneobject:summary(),plot(),as.data.frame()
-
Plus some more specific
metagenefunctions:b.values(),get.ntraits(),ngenerations(),nindividuals(),get.pedigree()
-
And specific functions about spatial arrangement:
coordinates()extract coordinatessim.spatial()simulates some spatial autocorrelation
Simulation framework
-
The function
breedR.sample.phenotype()simulates datasets from all the model structures available in breedR -
Limitation: only one generation, with random matings of founders
-
See
?simulationfor details
Remote computation
If you have access to a Linux server through SSH, you can perform computations remotely
-
Take advantage of more memory or faster processors
-
Parallelize jobs
-
Free local resources while fitting models
-
See
?remotefor details
Package options
-
breedR features a list of configurable options
-
Use
breedR.setOption(...)for changing an option during the current sesion -
Set options permanently in the file
$HOME/.breedRrc -
see
?breedR.optionfor details
breedR.getOption()
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.