Nothing
README.md
In driveR: Prioritizing Cancer Driver Genes Using Genomics Data
 driveR: An R Package for Prioritizing Cancer Driver Genes Using Genomics Data
driveR: An R Package for Prioritizing Cancer Driver Genes Using Genomics Data




Cancer genomes contain large numbers of somatic alterations but few
genes drive tumor development. Identifying cancer driver genes is
critical for precision oncology. Most of current approaches either
identify driver genes based on mutational recurrence or using estimated
scores predicting the functional consequences of mutations.
driveR is a tool for personalized or batch analysis of genomics data
for driver gene prioritization by combining genomics information and
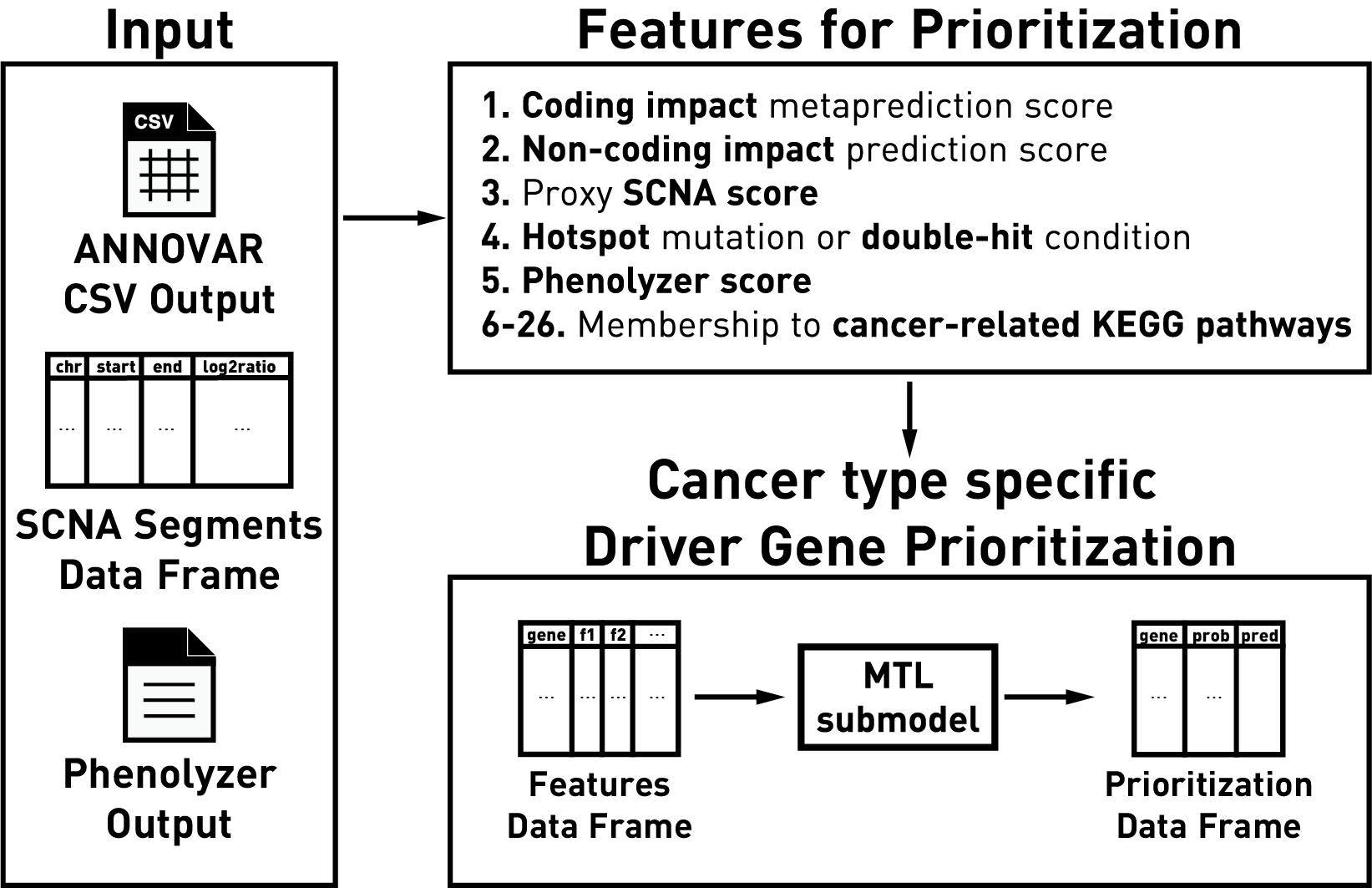
prior biological knowledge. As features, driveR uses coding impact
metaprediction scores, non-coding impact scores, somatic copy number
alteration scores, hotspot gene/double-hit gene condition, ‘phenolyzer’
gene scores and memberships to cancer-related KEGG pathways. It uses
these features to estimate cancer-type-specific probabilities for each
gene of being a cancer driver using the related task of a multi-task
learning classification model.
The method is described in detail in Ülgen E, Sezerman OU. driveR: a
novel method for prioritizing cancer driver genes using somatic genomics
data. BMC Bioinformatics. 2021 May
24;22(1):263.https://doi.org/10.1186/s12859-021-04203-7
Installation
You can install the latest released version of driveR from CRAN via:
install.packages("driveR")
You can install the development version of driveR from
GitHub with:
# install.packages("devtools")
devtools::install_github("egeulgen/driveR", build_vignettes = TRUE)
Usage
 driveR workflow
driveR workflow
driveR has two main objectives:
- Prediction of impact of coding variants (achieved via
predict_coding_impact())
- Prioritization of cancer driver genes (achieved via
create_features_df() and prioritize_driver_genes())
Note that driveR require operations outside of R and depends on the
outputs from the external tools ANNOVAR and phenolyzer.
For detailed information on how to use driveR, please see the vignette
“How to use driveR” via vignette("how_to_use")
Try the driveR package in your browser
Any scripts or data that you put into this service are public.
driveR documentation built on Aug. 19, 2023, 5:12 p.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
driveR: An R Package for Prioritizing Cancer Driver Genes Using Genomics Data
![]()
![]()
![]()
Cancer genomes contain large numbers of somatic alterations but few genes drive tumor development. Identifying cancer driver genes is critical for precision oncology. Most of current approaches either identify driver genes based on mutational recurrence or using estimated scores predicting the functional consequences of mutations.
driveR is a tool for personalized or batch analysis of genomics data
for driver gene prioritization by combining genomics information and
prior biological knowledge. As features, driveR uses coding impact
metaprediction scores, non-coding impact scores, somatic copy number
alteration scores, hotspot gene/double-hit gene condition, ‘phenolyzer’
gene scores and memberships to cancer-related KEGG pathways. It uses
these features to estimate cancer-type-specific probabilities for each
gene of being a cancer driver using the related task of a multi-task
learning classification model.
The method is described in detail in Ülgen E, Sezerman OU. driveR: a novel method for prioritizing cancer driver genes using somatic genomics data. BMC Bioinformatics. 2021 May 24;22(1):263.https://doi.org/10.1186/s12859-021-04203-7
Installation
You can install the latest released version of driveR from CRAN via:
install.packages("driveR")
You can install the development version of driveR from
GitHub with:
# install.packages("devtools")
devtools::install_github("egeulgen/driveR", build_vignettes = TRUE)
Usage
driveR workflow
driveR has two main objectives:
- Prediction of impact of coding variants (achieved via
predict_coding_impact()) - Prioritization of cancer driver genes (achieved via
create_features_df()andprioritize_driver_genes())
Note that driveR require operations outside of R and depends on the
outputs from the external tools ANNOVAR and phenolyzer.
For detailed information on how to use driveR, please see the vignette
“How to use driveR” via vignette("how_to_use")
Try the driveR package in your browser
Any scripts or data that you put into this service are public.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.