In isglobal-brge/dsOmicsClient: DataSHIELD client site Omics association functions.
```{css, echo=FALSE}
body .main-container {
max-width: 1280px !important;
}
body {
max-width: 1280px !important;
}
div.tocify {
width: 30%;
max-width: 310px;
}
tbody {
display: table-row-group;
vertical-align: middle;
border-color: inherit;
}
table tr {
background-color: #fff;
border-top: 1px solid #c6cbd1;
}
table tr:nth-child(2n) {
background-color: #f6f8fa;
}
table th {
font-weight: 600;
}
table td,
table th {
border: 1px solid #dfe2e5;
padding: 6px 13px;
}
table tr {
background-color: #fff;
border-top: 1px solid #c6cbd1;
}
table tr:nth-child(2n) {
background-color: #f6f8fa;
}
table > thead > tr > th {
border-bottom: 0;
}
```r
library(magrittr)
library(dplyr)
options(width = 80)
local({
hook_output <- knitr::knit_hooks$get('output')
knitr::knit_hooks$set(output = function(x, options) {
if (!is.null(options$max.height)) options$attr.output <- c(
options$attr.output,
sprintf('style="max-height: %s;"', options$max.height)
)
hook_output(x, options)
})
})
knitr::knit_hooks$set(inline = function(x) {
knitr:::format_sci(x, 'md')
})
knitr::opts_chunk$set(max.height='150px', cache=T, comment="", warning=FALSE, message=FALSE)
Introduction
Purpose
[...]
The functions to be executed in the local side (i.e. data analyst machine) will extremely facilitate the bioinformatic analyses by running simple R sentences for each of the required genomic data analysis. These functions are implemented in the dsOmicsClient package that includes:
- Principal component analyses (population stratification)
- Allele frequency
- Hardy-Weinberg equillibrium test
- Single association analyses (meta-analysis and mega-analysis, e.g pooled data)
- GWAS (meta-analysis)
- Polygenic risk score (PRS)
The package also allows to perform:
- Epigenome-wide association analyses
- Meta-analyses
- Post-omic data analyses (e.g. pathway/enrichment)
Use cases
Three different use cases will be illustrate:
- Single cohort GWAS. Using data from all the individuals.
- Multi-cohort GWAS. Using the same individuals as the single cohort separated into three synthetic cohorts.
- Polygenic risk score. Using data from all the individuals.
It's important noting that the data for the use cases is divided by chromosome (VCF files). It can be run by using a single VCF that contains all the variants, it is just not advisable from a performance point of view.
The data used along this section is a synthetic data set generated by the CINECA project made freely available under the Creative Commons Licence (CC-BY) (funding: EC H2020 grant 825775). It has been pruned and the individuals have been separated into three synthetic cohorts. For the "Single cohort" use case, all the individuals are used. Information on individuals and SNP count of each cohort can be found on the Table \@ref(tab:tab1).
| | Cohort 1 | Cohort 2 | Cohort 3 | Total |
|-----------------------|---------:|---------:|---------:|--------:|
| Number of SNPs | 865,240 | 865,240 | 865,240 | 865,240 |
| Number of individuals | 817 | 1,073 | 614 | 2,504 |
: (#tab:tab1) Numer of SNPs and individuals by cohort
Single cohort
The single cohort analysis is a way of performing a GWAS study guaranteeing GDPR data confidentiality. The structure followed is illustrated on the following figure.
knitr::include_graphics("gwas_images/single_cohort.png")
The data analyst corresponds to the "RStudio" session, which through DataSHIELD Interface (DSI) connects with the Opal server located at the cohort network. This Opal server contains an array of resources that correspond to the different VCF files (sliced by chromosome) [^1] and a resource that corresponds to the phenotypes table of the studied individuals.
[^1]: The same methodology and code can be used with unitary VCF resources that contain the variant information of all chromosomes.
Connection to the Opal server
We have to create an Opal connection object to the cohort server. We do that using the following functions.
require('DSI')
require('DSOpal')
require('dsBaseClient')
require('dsOmicsClient')
builder <- DSI::newDSLoginBuilder()
builder$append(server = "cohort1", url = "https://opal-demo.obiba.org/",
user = "dsuser", password = "password",
driver = "OpalDriver", profile = "omics")
logindata <- builder$build()
conns <- DSI::datashield.login(logins = logindata)
Assign the VCF resources
Now that we have created a connection object to the Opal, we have started a new R session on the server, our analysis will take place in this remote session, so we have to load the data into it. This is done via a two step process:
- Load the resources to the R session. The resources can be imagined as a data structure that contains the information about were to find a data set and the access credentials to it; we as users are not able to look at this information (it is privately stored on the Opal server), but we can load it into our remote R session to make use of it. Following that, the second step comes naturally.
- Resolve the resources. Once we have the information on how to access the data, we have to actually retrieve it on the remote R session to analyze it.
In this use case we will use 21 different resources from the GWAS project hosted on the demo Opal server. This resources correspond to VCF files with information on individual chromosomes. The names of the resources are chrXXA (where XX is the chromosome number). Following the Opal syntax, we will refer to them using the string GWAS.chrXXA.
To load the resources we will use the DSI::datashield.assign.resource() function. Note that along the use case we use the lapply function to simplify our code since we have to perform repetitive operations. lapply is a way of creating loops in R, for more information visit the documentation.
lapply(1:21, function(x){
DSI::datashield.assign.resource(conns, paste0("chr", x), paste0("GWAS.chr", x))
})
Now we have assigned all the resources named GWAS.chrXXA into our remote R session. We have assigned them to the variables called chrXX. To verify this step has been performed correctly, we could use the ds.class function to check for their class and that they exist on the remote session.
ds.class("chr1")
To resolve the resources and retrieve the data in the remote session we will use the DSI::datashield.assign.expr() function. This function runs a line of code on the remote session [^2], and in particular we want to run the function as.resource.object(), which is the DataSHIELD function in charge of resolving the resources.
[^2]: The same methodology and code can be used with unitary VCF resources that contain the variant information of all chromosomes.
lapply(1:21, function(x){
DSI::datashield.assign.expr(conns = conns, symbol = paste0("gds", x, "_object"),
expr = as.symbol(paste0("as.resource.object(chr", x, ")")))
})
Now we have resolved the resources named chrXX into our remote R session. The objects retrieved have been assigned into variables named gdsXX_object. We can check the process was successful as we did before.
ds.class("gds1_object")
Assign the phenotypes
The objects we have loaded into our remote session are VCF files that contain genomic information of the individuals. To perform a GWAS this information has to be related to some phenotypes to extract relationships. Therefore, we need to load the phenotypes into the remote session. The phenotypes information is a table that contains the individuals as rows and phenotypes as columns. In this use case, we will use a resource (as with the VCF files) to load the phenotypes table into the remote session.
The procedure is practically the same as before with some minor tweaks. To begin, we have the phenotypes information in a single table (hence, a single resource). Then, instead of using the function as.resource.object, we will use as.resource.data.frame, this is because before we were loading a special object (VCF file) and now we are loading a plain table, so the internal treatment on the remote session has to be different.
DSI::datashield.assign.resource(conns, "pheno", "GWAS.ega_phenotypes")
DSI::datashield.assign.expr(conns = conns, symbol = "pheno_object",
expr = quote(as.resource.data.frame(pheno)))
We can follow the same analogy as before to know that we have assigned the phenotypes table to a variable called pheno_object on the remote R session.
ds.class("pheno_object")
We can also check the column names to see which information is present on the table.
ds.colnames("pheno_object")[[1]]
Merge VCF (genotype) and phenotype information {#vcf_merge}
Arrived at this point, we have 21 VCF objects (each one corresponds to a chromosome) and a phenotypes table on our remote session. The next step is merging each of the VCF objects with the phenotypes table. Before doing that however, we have to gather some information from the phenotypes table. The information to gather is summarized on the Table \@ref(tab:tab2).
+---------------------------------------------------------------+----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------+
| Information | Details |
+===============================================================+====================================================================================================================================================================================================================================================+
| Which column has the samples identifier? | Column name that contains the IDs of the samples. Those are the IDs that will be matched to the ones present on the VCF objects. |
+---------------------------------------------------------------+----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------+
| Which is the sex column on the covariates file? | Column name that contains the sex information. |
+---------------------------------------------------------------+----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------+
| How are males and how are females encoded into this column? | There is not a standard way to encode the sex information, some people use 0/1; male/female; etc. Our approach uses a library that requires a specific encoding, for that reason we need to know the original encoding to perform the translation. |
+---------------------------------------------------------------+----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------+
| Which is the case-control column? | Column name that contains the case/control to be studied. |
+---------------------------------------------------------------+----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------+
| What is the encoding for case and for control on this column? | Similar as the sex column, the case/control will be translated to the particular standard of the software we have used to develop our functionalities [^3] . |
+---------------------------------------------------------------+----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------+
: (#tab:tab2) Information to gather from the phenotypes table.
[^3]: It's important to note that all the other encodings present on the case-control column that are not the case or control, will be turned into missings. As an example, if we have "cancer"/"no cancer"/"no information"/"prefer not to answer" we can specifcy case="cancer", control="no cancer" and all the individuals with "no information"/"prefer not to answer" will be interpreted as missings.
If we are not sure about the exact column names, we can use the function ds.colnames as we did before. Also, we can use the function ds.table1D to check the level names of the categorical variables.
ds.table1D("pheno_object$sex")$counts
ds.table1D("pheno_object$diabetes_diagnosed_doctor")$counts
From the different functions we used, we can extract our answers (Summarized on the Table \@ref(tab:tab3))
+-----------------------------------------------------------------+-----------------------------+
| Question | Answer |
+=================================================================+=============================+
| Which column has the samples identifier? | "subject_id" |
+-----------------------------------------------------------------+-----------------------------+
| Which is the sex column on the covariates file? | "sex" |
+-----------------------------------------------------------------+-----------------------------+
| How are males and how are females encoded into this column? | "male"/"female" |
+-----------------------------------------------------------------+-----------------------------+
| Which is the case-control column of interest of the covariates? | "diabetes_diagnosed_doctor" |
+-----------------------------------------------------------------+-----------------------------+
| What is the encoding for case and for control on this column? | "Yes"/"No" |
+-----------------------------------------------------------------+-----------------------------+
: (#tab:tab3) Summary of options from the example phenotypes table.
With all this information we can now merge the phenotypes and VCF objects into a type of object named GenotypeData. We will use the ds.GenotypeData function.
lapply(1:21, function(x){
ds.GenotypeData(x=paste0('gds', x,'_object'), covars = 'pheno_object', columnId = "subject_id",
sexId = "sex", male_encoding = "male", female_encoding = "female",
case_control_column = "diabetes_diagnosed_doctor", case = "Yes", control = "No",
newobj.name = paste0('gds.Data', x), datasources = conns)
})
The objects we just created are named gds.DataXX on the remote session. Now we are ready to perform a GWAS analysis. To perform a GWAS we have to supply this "Genotype Data" objects and some sort of formula in which we will specify the type of association we are interested on studying. The R language has it's own way of writing formulas, a simple example would be the following (relate the condition to the smoke variable adjusted by sex):
condition ~ smoke + sex
For this use case we will use the following formula.
diabetes_diagnosed_doctor ~ sex + hdl_cholesterol
Performing the GWAS
We are finally ready to achieve our ultimate goal, performing a GWAS analysis. We have our data (genotype + phenotypes) organized into the correspondent objects (GenotypeData) and our association formula ready. The function ds.metaGWAS is in charge of doing the analysis, we have to supply it with the names of the GenotypeData objects of the remote R session and the formula. Since we have 21 different objects, the paste0 function is used to simplify the call.
results_single <- ds.metaGWAS(genoData = paste0("gds.Data", 1:21), model = diabetes_diagnosed_doctor ~ sex + hdl_cholesterol)[[1]]
results_single
We can display the results of the GWAS using a Manhattan plot.
manhattan(results_single)
Moreover, we can visualize the region with the lowest p-value in detail using the function LocusZoom. There are multiple arguments to control the gene annotation, display a different region of the GWAS, display a different range of positions and more. Make sure to check ?LocusZoom for all the details.
LocusZoom(results_single, use_biomaRt = FALSE)
datashield.logout(conns)
Multi cohorts
Now all the individuals used for the single cohort use case will be divided into three different synthetic cohorts. The results from each cohort will then be meta-analyzed to be compared to the results obtained by studying all the individuals together. As with the single-cohort methodology illustrated on the prior section, this method guarantees GDPR data confidentiality. The structure for a three cohort study is illustrated on the following figure (note this can be extrapolated for cohorts with a bigger (or lower) partner count).
knitr::include_graphics("gwas_images/multi_cohort.png")
The data analyst corresponds to the "RStudio" session, which through DataSHIELD Interface (DSI) connects with the Opal server located at the cohort network. This Opal server contains an array of resources that correspond to the different VCF files (sliced by chromosome) [^4] and a resource that corresponds to the phenotypes table of the studied individuals.
[^4]: The same methodology and code can be used with unitary VCF resources that contain the variant information of all chromosomes.
This use case will portray a complete study methodology, more precisely we will follow this flowchart:
knitr::include_graphics("gwas_images/multi_cohort_flowchart.png")
Brief description of each step:
- Step 1: Logging into the Opal servers and load all the VCF files to the remote R sessions. Corresponds to the sections Connection to the Opal server, Assign the VCF resources, Assign the phenotypes and Merge VCF (genotype) and phenotype information.
- Step 2: Performing a GWAS analysis on each cohort and collect all the aggregated non-disclosive statistics. Corresponds to the sections Performing the GWAS and [GWAS Plots].
- Step 3: Apply different QC control methodologies to assess possible problems on the data used on each cohort to perform the analysis. Corresponds to the section [Meta-level quality control].
- Step 4: Perform the meta-analysis of the aggregated statistics. Corresponds to the section Meta-analysis.
Connection to the Opal server
We have to create an Opal connection object to the different cohorts server. We do that using the following functions.
require('DSI')
require('DSOpal')
require('dsBaseClient')
require('dsOmicsClient')
builder <- DSI::newDSLoginBuilder()
builder$append(server = "cohort1", url = "https://opal-demo.obiba.org/",
user = "dsuser", password = "password",
driver = "OpalDriver", profile = "omics")
builder$append(server = "cohort2", url = "https://opal-demo.obiba.org/",
user = "dsuser", password = "password",
driver = "OpalDriver", profile = "omics")
builder$append(server = "cohort3", url = "https://opal-demo.obiba.org/",
user = "dsuser", password = "password",
driver = "OpalDriver", profile = "omics")
logindata <- builder$build()
conns <- DSI::datashield.login(logins = logindata)
It is important to note that in this use case, we are only using one server that contains all the resources (https://opal-demo.obiba.org/), on this server there are all the resources that correspond to the different cohorts.
Assign the VCF resources {#multi-vcf}
Now that we have created a connection object to the different Opals, we have started three R session, our analysis will take place on those remote sessions, so we have to load the data into them.
In this use case we will use 63 different resources from the GWAS project hosted on the demo Opal server. This resources correspond to VCF files with information on individual chromosomes (21 chromosomes per three cohorts). The names of the resources are chrXXY (where XX is the chromosome number and Y is the cohort A/B/C). Following the Opal syntax, we will refer to them using the string GWAS.chrXXY.
We have to refer specifically to each different server by using conns[X], this allows us to communicate with the server of interest to indicate to it the resources that it has to load.
# Cohort 1 resources
lapply(1:21, function(x){
DSI::datashield.assign.resource(conns[1], paste0("chr", x), paste0("GWAS.chr", x,"A"))
})
# Cohort 2 resources
lapply(1:21, function(x){
DSI::datashield.assign.resource(conns[2], paste0("chr", x), paste0("GWAS.chr", x,"B"))
})
# Cohort 3 resources
lapply(1:21, function(x){
DSI::datashield.assign.resource(conns[3], paste0("chr", x), paste0("GWAS.chr", x,"C"))
})
Now we have assigned all the resources named GWAS.chrXXY into our remote R session. We have assigned them to the variables called chrXX. To verify this step has been performed correctly, we could use the ds.class function to check for their class and that they exist on the remote sessions.
ds.class("chr1")
We can see that the object chr1 exists in all the three servers.
Finally the resources are resolved to retrieve the data in the remote sessions.
lapply(1:21, function(x){
DSI::datashield.assign.expr(conns = conns, symbol = paste0("gds", x, "_object"),
expr = as.symbol(paste0("as.resource.object(chr", x, ")")))
})
Now we have resolved the resources named chrXX into our remote R sessions. The objects retrieved have been assigned into variables named gdsXX_object. We can check the process was successful as we did before.
ds.class("gds1_object")
Assign the phenotypes {#assign-the-phenotypes-1}
The objects we have loaded into our remote sessions are VCF files that contain genomic information of the individuals. To perform a GWAS this information has to be related to some phenotypes to extract relationships. Therefore, we need to load the phenotypes into the remote sessions. The phenotypes information is a table that contains the individuals as rows and phenotypes as columns. In this use case, we will use a resource (as with the VCF files) to load the phenotypes table into the remote sessions.
As with the VCF resources, here we load a different resource to each cohort, as it is expected that each cohort has only the phenotypes information regarding their individuals, therefore different tables. Note that we are assigning the resource to the same variable at each server (pheno), so we only need one function call to resolve all the resources together.
# Cohort 1 phenotypes table
DSI::datashield.assign.resource(conns[1], "pheno", "GWAS.ega_phenotypes_1")
# Cohort 2 phenotypes table
DSI::datashield.assign.resource(conns[2], "pheno", "GWAS.ega_phenotypes_2")
# Cohort 3 phenotypes table
DSI::datashield.assign.resource(conns[3], "pheno", "GWAS.ega_phenotypes_3")
# Resolve phenotypes table
DSI::datashield.assign.expr(conns = conns, symbol = "pheno_object",
expr = quote(as.resource.data.frame(pheno)))
We can follow the same analogy as before to know that we have assigned the phenotypes table to a variable called pheno_object on the remote R session.
ds.class("pheno_object")
We can also check the column names to see which information is present on the table.
ds.colnames("pheno_object")[[1]]
Merge VCF (genotype) and phenotype information {#merge-vcf-genotype-and-phenotype-information}
Arrived at this point, we have 21 VCF objects at each cohort R session (each one corresponds to a chromosome) and a phenotypes table. The next step is merging each of the VCF objects with the phenotypes table. The same procedure as the single cohort can be applied to extract the arguments required.
With all this information we can now merge the phenotypes and VCF objects into a type of object named GenotypeData. We will use the ds.GenotypeData function.
lapply(1:21, function(x){
ds.GenotypeData(x=paste0('gds', x,'_object'), covars = 'pheno_object', columnId = "subject_id", sexId = "sex",
male_encoding = "male", female_encoding = "female",
case_control_column = "diabetes_diagnosed_doctor", case = "Yes", control = "No",
newobj.name = paste0('gds.Data', x), datasources = conns)
})
The objects we just created are named gds.DataXX on the remote session. Now we are ready to perform a GWAS analysis. We will use the same formula as the single cohort to be able to compare the results later:
diabetes_diagnosed_doctor ~ sex + hdl_cholesterol
Performing a meta - GWAS {#performing-the-gwas-1 .tabset .tabset-fade .tabset-pills}
We now proceed to perform the GWAS analysis.
results <- ds.metaGWAS(genoData = paste0("gds.Data", 1:21), model = diabetes_diagnosed_doctor ~ sex + hdl_cholesterol)
And we can see the results for each cohort.
Cohort 1 {.unnumbered}
results[[1]]
Cohort 2 {.unnumbered}
results[[2]]
Cohort 3 {.unnumbered}
results[[3]]
GWAS Plots {.tabset .tabset-fade .tabset-pills}
We can display the results of the GWAS using a Manhattan plot for each cohort.
Cohort 1 {.unnumbered}
manhattan(results[[1]])
Cohort 2 {.unnumbered}
manhattan(results[[2]])
Cohort 3 {.unnumbered}
manhattan(results[[3]])
Meta-level quality control
Up to this point we have collected the aggregated statistics from each cohort and we have visualized them. Before meta-analyzing this statistics we have to perform some quality control to make sure there are no major issues. There are three different methods bundled in dsOmicsClient to perform such task:
- EAF Plot: Plotting the reported EAFs against a reference set, such as from the HapMap or 1000 Genomes projects, or from one specific study, can help visualize patterns that pinpoint strand issues, allele miscoding or the inclusion of individuals whose self-reported ancestry did not match their genetic ancestry.
- P-Z Plot: Plot to reveal issues with beta estimates, standard errors and P values. The plots compare P values reported in the association GWAS with P values calculated from Z-statistics (P.ztest) derived from the reported beta and standard error. A study with no issues should show perfect concordance. It will generate a plot per study, where concordance of all the study SNPs will be displayed.
- SE-N Plot: Plot to reveal issues with trait transformations. A collection of studies with no issues will fall on a line.
If any of these QC raises some inconsistencies, please contact the correspondent study analyst.
EAF Plot
For this plot we are comparing the estimated allele frequencies to a reference set, so the first step is to get ourselves a reference set. For this example we will be using the ALL.wgs.phase1_release_v3.20101123 reference set. We will download it and load it into the client R session.
reference <- readr::read_tsv("https://homepages.uni-regensburg.de/~wit59712/easyqc/exomechip/ALL.wgs.phase1_release_v3.20101123.snps_indels_sv.sites.EUR_AF.txt.exm.tab.sexChr.DI.txt.GZ")
reference
Now we just use the function eafPlot and we pass to it the GWAS results and the reference set.
eafPlot(x = results, reference = reference, rs_reference = "Rs", freq_reference = "Freq")
The obtained results are the expected, which is a linear correlation of slope one. This indicates the alleles are correctly encoded. For further explanation on how to assess the results produced by this plot, please refer to the Figure 4 of @winkler2014quality.
P-Z Plot
This plot will assess the issues with beta estimates, standard errors and P values. Here we will only need the results from the ds.GWAS function and pass them to the function pzPlot.
pzPlot(results)
Please note that all the SNPs are plotted, for that reason there may be some performance issues if there are many SNPs to be plotted.
A dataset with no issues will show perfect concordance. For further details please refer to the Figure 3 of @winkler2014quality.
SE-N Plot
Finally, we will assess issues with trait transformations. For this we will only need the results from the ds.GWAS function and pass them to the function seNPlot.
seNPlot(results)
If all the study points fall on a line, no issues are present. For further detail please refer to the Figure 2 of @winkler2014quality.
Meta-analysis
Given no problems have been detected on the meta-level QC, we will proceed. Up to this point, we have obtained association results for each cohort on the study. The next step is to combine this information using meta-analysis methods to derive a pooled estimate closest to the common truth. Each researcher might have an already built pipeline to do so, or a preferred method; nevertheless, we included a couple methods inside dsOmicsClient. They are the following:
- Meta-analysis of p-values: Using the sum of logs method (Fisher's method).
- Meta-analysis of beta values: Using a fixed-effects model. Methodology extracted and adapted from @betameta.
Assessment of the meta-analysis methods
Previously, we have calculated the same GWAS analysis both using all the individuals and a simulated three-cohort scenario. This gives us the ability to assess how good or bad are the different meta-analysis methods on discovering the closest common truth; to do so, we will compare both meta-analysis to the common truth, the evaluation method will be counting the number of top hits that match the common truth, more specifically we will search for matches on the top twenty hits.
We apply both methods to the multi-cohort use case results:
meta_pvalues <- metaPvalues(results)
meta_bvalues <- metaBetaValues(results)
We can now compare the 20 top hits from the common truth (previously stored on the variable results_single) to the 20 top hits yielded by both meta-analysis methodologies.
# Extract 20 top hits for all three cases
# All individuals
top_20_complete <- results_single %>% arrange(p.value) %>% head(20)
# Pvalue meta-analysis
top_20_pval <- meta_pvalues %>% arrange(p.meta) %>% head(20)
# Beta meta-analysis
top_20_beta <- meta_bvalues %>% arrange(pval) %>% head(20)
# Number of hits shared by all individuals case and beta meta-analysis
sum(top_20_complete$rs %in% top_20_beta$rs)
# Number of hits shared by all individuals case and pvalue meta-analysis
sum(top_20_complete$rs %in% top_20_pval$rs)
The meta-analysis using beta values recovers a higher amount of top SNPs, therefore it can be considerated a more powerful method for GWAS meta-analyses.
Visualizing the results of the meta-analysis
We can use the manhattan plot and locus zoom to visualize the results of the meta-analysis as we visualized the single cohorts before. To help assess the meta-analysis behaviour, we will plot together the meta-analysis results (left plot) and the common truth results (right plot).
Manhatan plot {.tabset .tabset-fade .tabset-pills}
Meta-analysis {.unnumbered}
manhattan(meta_bvalues)
Common truth {.unnumbered}
manhattan(results_single)
LocusZoom {.tabset .tabset-fade .tabset-pills}
Meta-analysis {#meta-analysis-2 .unnumbered}
LocusZoom(meta_bvalues, pvalue = "p.F", use_biomaRt = FALSE)
Common truth {.unnumbered}
LocusZoom(results_single, use_biomaRt = FALSE)
Meta-analysis versus pooled linear models
The dsOmicsClient package also bundles a function to fit pooled generalized linear models (GLM) on single (or multiple) SNPs. On this section we will compare the results of the meta-analysis to the pooled GLM models to see which method is closer to the common truth. The meta-analysis method will be the beta values (fixed-effects model).
The study methodology will be similar to the one we just performed. The top 20 hits yielded by the meta-analysis will be selected, those SNPs will be studied using the pooled GLM function. Both results will be compared to the common truth.
We aim to replicate a real scenario with this study, typically there is no access to the common truth (if so there is no point on performing meta-analysis on synthetic multi-cohorts), therefore we simulate the discovery of SNPs of interest via meta-analysis, then we want to assess if studying individually those SNPs using pooled GLM will put us closer to the common truth than what we already achieved.
We have already calculated the top 20 SNPs discovered by the beta meta-analysis.
top_20_beta
Next, we have to fit pooled GLM using the ds.glmSNP function, obviously we have to fit the same model that we did on the GWAS (Diabetes.diagnosed.by.doctor ~ sex + HDL.cholesterol), otherwise the results will not be consistent. Remember that we have the genotype data separated by chromosome, following that we will construct the GLM calls as follows:
s <- lapply(1:nrow(top_20_beta), function(x){
tryCatch({
ds.glmSNP(snps.fit = top_20_beta[x, 1], model = diabetes_diagnosed_doctor ~ sex + hdl_cholesterol, genoData = paste0("gds.Data", top_20_beta[x, 2]))
}, error = function(w){NULL})
})
# Simple data cleaning and column name changes
ss <- data.frame(do.call(rbind, s))
ss <- ss %>% rownames_to_column("rs")
pooled <- ss %>% dplyr::rename(Est.pooled = Estimate, SE.pooled = Std..Error, pval.pooled = p.value) %>% dplyr::select(-c(n, p.adj))
# ds.glmSNP(snps.fit = unlist(top_20_beta[3:5, 1]), model = diabetes_diagnosed_doctor ~ sex + hdl_cholesterol, genoData = paste0("gds.Data", unlist(top_20_beta[3:5, 2])), mc.cores = 3)
We now extract the results of interest from the meta-analysis and the common truth.
# Get data from all individuals study
original <- results_single %>% dplyr::rename(pval.original = p.value, Est.original = Est, SE.original = Est.SE) %>%
dplyr::select(c(rs, Est.original, SE.original, pval.original))
# Arrange dadta from beta meta-analysis
beta <- top_20_beta %>% dplyr::rename(pval.beta = pval, Est.beta = b.F, SE.beta = se.F) %>%
dplyr::select(c(rs, Est.beta, SE.beta, pval.beta))
# Merge all data on a single table
total <- Reduce(function(x,y){inner_join(x,y, by = "rs")}, list(original, pooled, beta))
With all the information gathered we can visualize the results on the Table 4.
[Table 4: Beta values, standard errors and p-values yielded by the three methods: GWAS of all individuals, meta-analysis of synthetic cohorts, pooled analysis of synthetic cohorts.]{#tab:tab4 style="font-size:8pt"}
library(gt)
total %>% gt() %>% tab_spanner(label = 'Beta', columns = c(Est.original, Est.pooled, Est.beta)) %>% tab_spanner(label = 'SE', columns = c(SE.original, SE.pooled, SE.beta)) %>% tab_spanner(label = 'P-Value', columns = c(pval.original, pval.pooled, pval.beta)) %>% cols_label(Est.original = "Original", Est.pooled = "Pooled", Est.beta = "Meta", SE.original = "Original", SE.pooled = "Pooled", SE.beta = "Meta", pval.original = "Original", pval.pooled = "Pooled", pval.beta = "Meta", rs = "SNP id") %>% fmt_number(columns = c(Est.original, Est.pooled, Est.beta, SE.original, SE.pooled, SE.beta), decimals = 3) %>% fmt_scientific(columns = c(pval.original, pval.pooled, pval.beta), decimals = 1) %>% tab_options(column_labels.font.weight = "bold") %>% tab_style(style = cell_fill(color = "grey"), locations = list(cells_body(columns = c(SE.original, SE.pooled, SE.beta), rows = c(seq(2,nrow(total),2))))) %>% tab_style(style = cell_fill(color = "lightgray"), locations = list(cells_body(columns = c(SE.original, SE.pooled, SE.beta), rows = c(seq(1,nrow(total),2))))) %>% tab_style(style = cell_borders(sides = "all", color = "lightgrey", style = "solid", weight = px(1)), locations = cells_body(columns = everything(), rows = everything())) %>% tab_style(style = cell_text(color = "red"), locations = list(cells_body(columns = Est.original, rows = Est.original<0))) %>% tab_style(style = cell_text(color = "red"), locations = list(cells_body(columns = Est.beta, rows = Est.beta<0))) %>% tab_style(style = cell_text(color = "red"), locations = list(cells_body(columns = Est.pooled, rows = Est.pooled<0))) %>% tab_options(table.font.size = "10pt")
Finally, to conclude this study we can compute the bias and mean squared error (code not shown). The results are shown on the Table 5.
# MSE
mse_est_pooled <- mean((total$Est.original - total$Est.pooled)^2)
mse_est_beta <- mean((total$Est.original - total$Est.beta)^2)
# Bias
bias_est_pooled <- mean(total$Est.original - total$Est.pooled)
bias_est_beta <- mean(total$Est.original - total$Est.beta)
statistics <- data.frame(MSE = c(mse_est_pooled, mse_est_beta), Bias = c(bias_est_pooled, bias_est_beta), stat = c("Beta", "Beta"), var = c("Pooled", "Meta"))
[Table 5: Mean squared error and bias for the betas from the meta-analysis and pooled analysis compared to the common truth (all individuals).]{#tab:tab5 style="font-size:8pt"}
statistics %>% group_by(stat) %>% gt(rowname_col = "var") %>% fmt_scientific(columns = c(MSE, Bias), decimals = 1) %>% tab_options(column_labels.font.weight = "bold") %>% tab_options(table.font.size = "10pt")
With this results we can conclude that refining the statistics for the SNPs of interest using pooled GLM is advisable. Therefore the recommended flowchart when performing a GWAS using dsOmicsClient+DataSHIELD is the following:
REVISAR ESTOS RESULTADOS
- Perform GWAS with
dsOmicsClient::dsGWAS
- Perform meta-analysis on the results with
dsOmicsClient::metaBetaValues
- Extract top hits from the meta-analysis.
- Perform pooled GLM on top hits with
dsOmicsClient::ds.glmSNP
Fast GWAS analysis
An alternate methodology to performing a meta analysis is to use the fast GWAS methodology. This method is named after the performance it achieves, however, it does provide further advantages other than computational speed. The main advantage is that it does not compute the results for each cohort to then be meta-analyzed, it actually computes them using a pooled technique that achieves the exact same results when all the individuals are together than when they are separated by cohorts. This is of great importance as there is no need to worry about cohort-imbalance, which is usually a problem in multi-cohort meta-analysis studies. The only downside of this method is that it does not yield the same results of a traditional GWAS analysis pipeline, the inacuraccy of the results however is really low, detailed information about that can be found on the paper that proposes the method.
The usage is similar to the meta-GWAS with a couple of added options, feel free to check the documentation with ?ds.fastGWAS to see information about them.
results_fast <- ds.fastGWAS(genoData = paste0("gds.Data", 1:21), formula = "diabetes_diagnosed_doctor ~ sex + hdl_cholesterol", family = "binomial", snpBlock = 5000)
datashield.logout(conns)
GWAS Plot
We can display the results of the fast GWAS using a Manhattan plot.
manhattan(results_fast, pvalCol = 4)
Meta-analysis versus fast GWAS
To finish the GWAS section we can evaluate the results of the beta meta-analysis and the results of the fast GWAS. Both methods will be compared to the ground truth.
As before we can start by seeing the amount of SNPs that both methodologies recover as top hits.
# Extract 20 top hits for all three cases
# All individuals
top_20_complete <- results_single %>% arrange(p.value) %>% head(20)
# Fast GWAS
top_20_fast <- results_fast %>% arrange(p.value) %>% head(20)
# Beta meta-analysis
top_20_beta <- meta_bvalues %>% arrange(pval) %>% head(20)
# Number of hits shared by all individuals case and beta meta-analysis
sum(top_20_complete$rs %in% top_20_beta$rs)
# Number of hits shared by all individuals case and fast GWAS
sum(top_20_complete$rs %in% top_20_fast$rs)
On that regard it seems the meta-analysis is performing better.
We now extract the results of interest from the meta-analysis and the common truth.
# Get data from all individuals study
original <- results_single %>% dplyr::rename(pval.original = p.value, Est.original = Est, SE.original = Est.SE) %>%
dplyr::select(c(rs, Est.original, SE.original, pval.original))
# Arrange dadta from beta meta-analysis
beta <- top_20_beta %>% dplyr::rename(pval.beta = pval, Est.beta = b.F, SE.beta = se.F) %>%
dplyr::select(c(rs, Est.beta, SE.beta, pval.beta))
# Arrange data from fast GWAS
fast <- top_20_fast %>% dplyr::rename(pval.fast = p.value, Est.fast = Est, SE.fast = Est.SE) %>%
dplyr::select(c(rs, Est.fast, SE.fast, pval.fast))
# Merge all data on a single table
total <- Reduce(function(x,y){inner_join(x,y, by = "rs")}, list(original, fast, beta))
With all the information gathered we can visualize the results on the Table 6.
[Table 6: Beta values, standard errors and p-values yielded by the three methods: GWAS of all individuals, meta-analysis of synthetic cohorts, fast GWAS.]{#tab:tab6 style="font-size:8pt"}
library(gt)
total %>% gt() %>% tab_spanner(label = 'Beta', columns = c(Est.original, Est.fast, Est.beta)) %>% tab_spanner(label = 'SE', columns = c(SE.original, SE.fast, SE.beta)) %>% tab_spanner(label = 'P-Value', columns = c(pval.original, pval.fast, pval.beta)) %>% cols_label(Est.original = "Original", Est.fast = "Fast", Est.beta = "Meta", SE.original = "Original", SE.fast = "Fast", SE.beta = "Meta", pval.original = "Original", pval.fast = "Fast", pval.beta = "Meta", rs = "SNP id") %>% fmt_number(columns = c(Est.original, Est.fast, Est.beta, SE.original, SE.fast, SE.beta), decimals = 3) %>% fmt_scientific(columns = c(pval.original, pval.fast, pval.beta), decimals = 1) %>% tab_options(column_labels.font.weight = "bold") %>% tab_style(style = cell_fill(color = "grey"), locations = list(cells_body(columns = c(SE.original, SE.fast, SE.beta), rows = c(seq(2,nrow(total),2))))) %>% tab_style(style = cell_fill(color = "lightgray"), locations = list(cells_body(columns = c(SE.original, SE.fast, SE.beta), rows = c(seq(1,nrow(total),2))))) %>% tab_style(style = cell_borders(sides = "all", color = "lightgrey", style = "solid", weight = px(1)), locations = cells_body(columns = everything(), rows = everything())) %>% tab_style(style = cell_text(color = "red"), locations = list(cells_body(columns = Est.original, rows = Est.original<0))) %>% tab_style(style = cell_text(color = "red"), locations = list(cells_body(columns = Est.beta, rows = Est.beta<0))) %>% tab_style(style = cell_text(color = "red"), locations = list(cells_body(columns = Est.fast, rows = Est.fast<0))) %>% tab_options(table.font.size = "10pt")
Finally, to conclude this study we can compute the bias and mean squared error (code not shown). The results are shown on the Table 7.
# MSE
mse_est_fast <- mean((total$Est.original - total$Est.fast)^2)
mse_est_beta <- mean((total$Est.original - total$Est.beta)^2)
# Bias
bias_est_fast <- mean(total$Est.original - total$Est.fast)
bias_est_beta <- mean(total$Est.original - total$Est.beta)
statistics <- data.frame(MSE = c(mse_est_fast, mse_est_beta), Bias = c(bias_est_fast, bias_est_beta), stat = c("Beta", "Beta"), var = c("Fast", "Meta"))
[Table 7: Mean squared error and bias for the betas from the meta-analysis and fast GWAS compared to the common truth (all individuals).]{#tab:tab5 style="font-size:8pt"}
statistics %>% group_by(stat) %>% gt(rowname_col = "var") %>% fmt_scientific(columns = c(MSE, Bias), decimals = 1) %>% tab_options(column_labels.font.weight = "bold") %>% tab_options(table.font.size = "10pt")
On this brief comparison we've seen that the fast GWAS retrieves sensibly less SNPs from the ground truth but it has better performance of bias and MSE. It must be noted that the data used for this GWAS examples does not suffer from any cohort imbalance, so the meta-analysis is expected to yield proper results, on a different environment the fast GWAS is expected to exceed the performance of the meta-analysis in a more remarkable manner.
Post omic analysis
Up to this point we have analyzed the genotype and phenotype data by performing a genome wide association study. We can further study the yielded results by performing an enrichment analysis.
Enrichment analysis
The GWAS results are a collection of SNPs that displayed a relationship with a given phenotype. Those SNPs may fall inside gene regions, and those genes can be functionally related. Studying this can help researchers understand the underlying biological processes. Those analysis are carried by querying the selected gene to a database, for this example we will be using the DisGeNET database.
Annotation of genes
The first step of the enrichment analysis is to annotate the genes that contain the SNPs of interest yielded by the GWAS. There are many ways to perform this step, we propose to use the biomaRt package and retrieve the annotation from the ensembl genome browser.
First we select the SNPs of interest. We have established a p.value threshold to do so.
snps_of_interest <- results_fast %>% dplyr::arrange(p.value) %>% dplyr::select(chr, pos, rs, p.value) %>% dplyr::filter(p.value < 0.00005)
And with this list of SNPs we can move to the annotation.
library(biomaRt)
# Perform ensembl connection
gene.ensembl <- useEnsembl(biomart = "ensembl", dataset = "hsapiens_gene_ensembl", GRCh = 37)
# Query SNP by SNP
genes_interest <- Reduce(rbind, apply(snps_of_interest, 1, function(x){
result <- getBM(
attributes = c('entrezgene_id', 'chromosome_name', 'ensembl_gene_id', 'start_position', "end_position"),
filters = c('start', 'end', 'chromosome_name'),
values = list(start = x[2], end = x[2], chromosome_name = x[1]),
mart = gene.ensembl)
}))
Enrichment analysis query
Having the genes of interest annotated, the final step is to query them to DisGeNET. We will use the function enrichDGN from the package DOSE for it. This package is part of a large ecosystem of packages dedicated to enrichment analysis, this allows for easy representation of the query results.
library(DOSE)
enrichment <- enrichDGN(unlist(genes_interest$entrezgene_id))
Enrichment analysis plots
Here we will portray some of the visualizations that can be performed with the query results. For more information and further visualization options please read the official vignette.
Barplot
# library(enrichplot)
# barplot(enrichment)
Dotplot
# dotplot(enrichment)
CNET plot
# library(ggnewscale)
# # convert gene ID to Symbol
# edox <- setReadable(enrichment, 'org.Hs.eg.db', 'ENTREZID')
# cnetplot(edox, foldChange=genes_interest$entrezgene_id)
Polygenic risk scores
Definition
By checking for specific variants and quantifying them, a researcher can extract the polygenic risk score (PRS) of an individual, which translates to an associated risk of the individual versus a concrete phenotype. A definition of the term reads
"A weighted sum of the number of risk alleles carried by an individual, where the risk alleles and their weights are defined by the loci and their measured effects as detected by genome wide association studies." (Extracted from @torkamani2018personal)
The use of PRS markers is very popular nowadays and it has been proved to be a good tool to asses associated risks @escott2017polygenic, @forgetta2020development, @kramer2020breast.
When calculating the PRS using DataSHIELD, the actual scores are not returned, as it would not be GDPR compliant to be able to determine the risk of an individual versus a certain disease. The actual results are stored on the servers and optionally can be merged to the phenotypes table. This allows further study of the PRS using summaries or fitting pooled GLM models to assess relationships between phenotypes and PRS scores.
Study configuration
The PRS use case will be set up as a multi-cohort, using the same dataset as the multi-cohort GWAS (same VCF resources). A single cohort PRS could be performed analogously using the single cohort example of the GWAS.
knitr::include_graphics("gwas_images/prs.png")
Source of polygenic scores
There are two ways of stating the SNPs of interest and their weights in order to calculate the PRS.
-
Providing a prs_table (SNPs of interest) table. The prs_table table has to have a defined structure and column names in order to be understood by this function. This table can be formulated using two schemas:
- Scheme 1: Provide SNP positions. Use the column names: "
chr_name", "chr_position", "effect_allele", "reference_allele", "effect_weight"
| chr_name | chr_position | effect_allele | reference_allele | effect_weight |
|----------|--------------|---------------|------------------|---------------|
| 1 | 100880328 | T | A | 0.0373 |
| 1 | 121287994 | G | A | -0.0673 |
| 6 | 152023191 | A | G | 0.0626 |
| ... | ... | ... | ... | ... |
- Scheme 2: Provide SNP id's. Use the column names: "
rsID", "effect_allele", "reference_allele", "effect_weight".
| rdID | effect_allele | reference_allele | effect_weight |
|------------|---------------|------------------|---------------|
| rs11206510 | T | C | 0.0769 |
| rs4773144 | G | A | 0.0676 |
| rs17514846 | A | C | 0.0487 |
| ... | ... | ... | ... |
The effect_weight have to be the betas (log(OR)).
- Provide a PGS Catalog 'Polygenic Score ID & Name'. If this option is in use, the SNPs and beta weights will be downloaded from the PGS Catalog and will be used to calculate the PRS.
Please note when using a custom prs_table table that it is much faster to use the Schema 1, as the subset of the VCF files is miles faster to do using chromosome name and positions rather than SNP id's.
Connection to the Opal server {#connection-to-the-opal-server-2}
We have to create an Opal connection object to the cohort server. We do that using the following functions.
require('DSI')
require('DSOpal')
require('dsBaseClient')
require('dsOmicsClient')
builder <- DSI::newDSLoginBuilder()
builder$append(server = "cohort1", url = "https://opal-demo.obiba.org/",
user = "dsuser", password = "password",
driver = "OpalDriver", profile = "omics")
builder$append(server = "cohort2", url = "https://opal-demo.obiba.org/",
user = "dsuser", password = "password",
driver = "OpalDriver", profile = "omics")
builder$append(server = "cohort3", url = "https://opal-demo.obiba.org/",
user = "dsuser", password = "password",
driver = "OpalDriver", profile = "omics")
logindata <- builder$build()
conns <- DSI::datashield.login(logins = logindata)
Assign the VCF resources
We assign the VCF and phenotypes resources.
# Cohort 1 resources
lapply(1:21, function(x){
DSI::datashield.assign.resource(conns[1], paste0("chr", x), paste0("GWAS.chr", x,"A"))
})
DSI::datashield.assign.resource(conns[1], "phenotypes_resource", "GWAS.ega_phenotypes_1")
# Cohort 2 resources
lapply(1:21, function(x){
DSI::datashield.assign.resource(conns[2], paste0("chr", x), paste0("GWAS.chr", x,"B"))
})
DSI::datashield.assign.resource(conns[2], "phenotypes_resource", "GWAS.ega_phenotypes_2")
# Cohort 3 resources
lapply(1:21, function(x){
DSI::datashield.assign.resource(conns[3], paste0("chr", x), paste0("GWAS.chr", x,"C"))
})
DSI::datashield.assign.resource(conns[3], "phenotypes_resource", "GWAS.ega_phenotypes_3")
And we resolve the phenotypes table. Remember to pass the table through the ds.make_valid_names function to be sure the column names have valid R syntax.
DSI::datashield.assign.expr(conns = conns, symbol = "phenotypes",
expr = as.symbol("as.resource.data.frame(phenotypes_resource)"))
ds.make_valid_names("phenotypes")
There is one crucial difference between this analysis and the GWAS, here we don't need to resolve the resources. This is for memory efficiency purposes. When we calculate a PRS, we are only interested in a small selection of SNPs, for that reason the (potentially) huge VCF files will be trimmed before being resolved, so only the relevant SNPs are loaded in the memory. For that reason the resolving of the resources is delegated to the ds.PRS function, which will perform this trimming according to the SNPs of interest yielded by the PGS catalog query or the prs_table table.
Calculate the PRS
The PRS results will be added to the phenotypes table to be later studied using a pooled GLM. To do that, the following arguments of the function ds.PRS will be used:
table: Name of the table where the results will be merged.table_id_column: Number of the column in the table table that contains the individuals ID to merge the results from the PRS.table_prs_name: Name to append to the PRS results. It will take the structure prs_table_prs_name and prs_nw_table_prs_name.
PGS Catalog Example
For this example we will calculate the PRS associated to the trait 'HDL Cholesterol' and 'Thyroid cancer', which corresponds to the PGS ID PGS000660 and PGS000636
resources <- paste0("chr", 1:21)
names(resources) <- paste0("chr", 1:21)
resources <- as.list(resources)
# HDL cholesterol
ds.PRS(resources = resources, pgs_id = "PGS000660", table = "phenotypes", table_id_column = "subject_id")
# Thyroid cancer
ds.PRS(resources = resources, pgs_id = "PGS000636", table = "phenotypes", table_id_column = "subject_id")
We can check that the results have been correctly added to the phenotypes table.
tail(ds.colnames('phenotypes')[[1]])
Custom prs_table example
Here we will illustrate a use case with a custom prs_table, please note that the proposed SNPs of interest and betas are totally arbitrary.
We will first assemble the prs_table table following the indications found on the Details section of ?ds.PRS (Schema 1).
prs_table <- data.frame(chr_name = c(9, 9, 9, 9, 1, 1, 2, 2),
chr_position = c(177034, 161674, 140859, 119681, 60351, 825410, 154629, 145710),
effect_allele = c("T", "C", "G", "C", "G", "G", "T", "A"),
reference_allele = c("G", "T", "A", "T", "A", "A", "G", "G"),
effect_weight = c(0.5, 0.2, 0.1, 0.2, 0.1, 0.3, 0.1, 0.8))
Now we can pass the custom prs_table to the PRS function to retrieve the aggregated results.
resources <- paste0("chr", 1:21)
names(resources) <- paste0("chr", 1:21)
resources <- as.list(resources)
ds.PRS(resources = resources, prs_table = prs_table, table = "phenotypes", table_id_column = "subject_id", table_prs_name = "custom_prs_table_results")
Study of the PRS results
We can fit pooled GLM models using the function ds.glm. In this use case we will fit a model to relate the HDL.cholesterol to prs_PGS000660 adjusted by sex. The same will be done with the prs_PGS000636.
ds.glm(formula = "hdl_cholesterol ~ prs_PGS000660 + sex", family = "gaussian", data = "phenotypes")$coefficients
ds.glm(formula = "hdl_cholesterol ~ prs_PGS000636 + sex", family = "gaussian", data = "phenotypes")$coefficients
datashield.logout(conns)
Technical details
GWAS
TODO
Internal structure of the PRS functionality
The PRS function can be divided into three main blocks that perform the required tasks to come up with the results. The three functional blocks are the following:
- Client: Assemble the
prs_table table if PGS Catalog is used, perform calls to the server to subset the resources and calculate the PRS with the subsetted resources.
- Server resolver: Resolve the selected resources (VCF files) and subset them using the prs_table.
- Server analysis: Use the resolved resources (small VCF files with only the SNPs of interest according to the
prs_table table) to calculate the PRS.
In order to introduce a little how those three blocks work internally, schematized flow charts have been designed. To understand the exact inner working of the functionality it is advised to follow the flowcharts alongside the actual source code. Find the links to the three blocks source code, client, server resolver and server analysis.
Client
The client performs two basic tasks. The first one is to select between a user introduced prs_table or to retrieve the prs_table from the PGS Catalog. Please note that prs_table stands for Region Of Interest, which may not be the common term for PGS analysis but it makes sense since it refers to a table that contain the SNP's of interest and their weight to the score. If the user introduces a custom prs_table and a PGS Catalog ID, only the introduced table will be used, discarding the PGS Catalog ID information. Once this table is assembled, the next step is to call the required functions on the study servers. First, the resource resolver is called, and after that, the function that calculates the PRS is called.
This is illustrated on the following figure.
knitr::include_graphics("gwas_images/block1.png")
Server resolver
The server resolver is in charge of resolving the resources. The interesting aspect is that only a region of interest is actually assigned to the R session, this is to avoid overloading the memory with unnecessary information. There are two different methodologies to perform this subsetting, one is using chromosome names and position and the other is using the SNP id's. Due to the technical specification of VCF files, is much easier to perform the subsetting using chromosome names and positions because there is an indexing file for the VCF files to perform fast queries of regions by position. On the other hand, to filter a VCF file using SNP id's, the whole file has to be scanned, yielding a much slower process.
This block is illustrated on the following figure.
knitr::include_graphics("gwas_images/block2.png")
Server analysis
Many processes are performed inside the analysis block. For that reason, more than a flow chart, a step by step guide has been illustrated with the objects that are created (or modified) on each step. The most important step on this block is making sure that the alternate (risk) alleles match between the VCF resources and the alternate alleles stated by the prs_table or PGS Catalog.
The block is illustrated on the following figure.
knitr::include_graphics("gwas_images/block3.png")
References
isglobal-brge/dsOmicsClient documentation built on March 20, 2023, 3:52 p.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
```{css, echo=FALSE} body .main-container { max-width: 1280px !important;
} body { max-width: 1280px !important;
}
div.tocify { width: 30%; max-width: 310px; }
tbody { display: table-row-group; vertical-align: middle; border-color: inherit; }
table tr { background-color: #fff; border-top: 1px solid #c6cbd1; }
table tr:nth-child(2n) { background-color: #f6f8fa; }
table th { font-weight: 600; }
table td, table th { border: 1px solid #dfe2e5; padding: 6px 13px; }
table tr { background-color: #fff; border-top: 1px solid #c6cbd1; }
table tr:nth-child(2n) { background-color: #f6f8fa; }
table > thead > tr > th { border-bottom: 0; }
```r library(magrittr) library(dplyr) options(width = 80) local({ hook_output <- knitr::knit_hooks$get('output') knitr::knit_hooks$set(output = function(x, options) { if (!is.null(options$max.height)) options$attr.output <- c( options$attr.output, sprintf('style="max-height: %s;"', options$max.height) ) hook_output(x, options) }) }) knitr::knit_hooks$set(inline = function(x) { knitr:::format_sci(x, 'md') }) knitr::opts_chunk$set(max.height='150px', cache=T, comment="", warning=FALSE, message=FALSE)
Introduction
Purpose
[...]
The functions to be executed in the local side (i.e. data analyst machine) will extremely facilitate the bioinformatic analyses by running simple R sentences for each of the required genomic data analysis. These functions are implemented in the dsOmicsClient package that includes:
- Principal component analyses (population stratification)
- Allele frequency
- Hardy-Weinberg equillibrium test
- Single association analyses (meta-analysis and mega-analysis, e.g pooled data)
- GWAS (meta-analysis)
- Polygenic risk score (PRS)
The package also allows to perform:
- Epigenome-wide association analyses
- Meta-analyses
- Post-omic data analyses (e.g. pathway/enrichment)
Use cases
Three different use cases will be illustrate:
- Single cohort GWAS. Using data from all the individuals.
- Multi-cohort GWAS. Using the same individuals as the single cohort separated into three synthetic cohorts.
- Polygenic risk score. Using data from all the individuals.
It's important noting that the data for the use cases is divided by chromosome (VCF files). It can be run by using a single VCF that contains all the variants, it is just not advisable from a performance point of view.
The data used along this section is a synthetic data set generated by the CINECA project made freely available under the Creative Commons Licence (CC-BY) (funding: EC H2020 grant 825775). It has been pruned and the individuals have been separated into three synthetic cohorts. For the "Single cohort" use case, all the individuals are used. Information on individuals and SNP count of each cohort can be found on the Table \@ref(tab:tab1).
| | Cohort 1 | Cohort 2 | Cohort 3 | Total | |-----------------------|---------:|---------:|---------:|--------:| | Number of SNPs | 865,240 | 865,240 | 865,240 | 865,240 | | Number of individuals | 817 | 1,073 | 614 | 2,504 |
: (#tab:tab1) Numer of SNPs and individuals by cohort
Single cohort
The single cohort analysis is a way of performing a GWAS study guaranteeing GDPR data confidentiality. The structure followed is illustrated on the following figure.
knitr::include_graphics("gwas_images/single_cohort.png")
The data analyst corresponds to the "RStudio" session, which through DataSHIELD Interface (DSI) connects with the Opal server located at the cohort network. This Opal server contains an array of resources that correspond to the different VCF files (sliced by chromosome) [^1] and a resource that corresponds to the phenotypes table of the studied individuals.
[^1]: The same methodology and code can be used with unitary VCF resources that contain the variant information of all chromosomes.
Connection to the Opal server
We have to create an Opal connection object to the cohort server. We do that using the following functions.
require('DSI') require('DSOpal') require('dsBaseClient') require('dsOmicsClient') builder <- DSI::newDSLoginBuilder() builder$append(server = "cohort1", url = "https://opal-demo.obiba.org/", user = "dsuser", password = "password", driver = "OpalDriver", profile = "omics") logindata <- builder$build() conns <- DSI::datashield.login(logins = logindata)
Assign the VCF resources
Now that we have created a connection object to the Opal, we have started a new R session on the server, our analysis will take place in this remote session, so we have to load the data into it. This is done via a two step process:
- Load the resources to the R session. The resources can be imagined as a data structure that contains the information about were to find a data set and the access credentials to it; we as users are not able to look at this information (it is privately stored on the Opal server), but we can load it into our remote R session to make use of it. Following that, the second step comes naturally.
- Resolve the resources. Once we have the information on how to access the data, we have to actually retrieve it on the remote R session to analyze it.
In this use case we will use 21 different resources from the GWAS project hosted on the demo Opal server. This resources correspond to VCF files with information on individual chromosomes. The names of the resources are chrXXA (where XX is the chromosome number). Following the Opal syntax, we will refer to them using the string GWAS.chrXXA.
To load the resources we will use the DSI::datashield.assign.resource() function. Note that along the use case we use the lapply function to simplify our code since we have to perform repetitive operations. lapply is a way of creating loops in R, for more information visit the documentation.
lapply(1:21, function(x){ DSI::datashield.assign.resource(conns, paste0("chr", x), paste0("GWAS.chr", x)) })
Now we have assigned all the resources named GWAS.chrXXA into our remote R session. We have assigned them to the variables called chrXX. To verify this step has been performed correctly, we could use the ds.class function to check for their class and that they exist on the remote session.
ds.class("chr1")
To resolve the resources and retrieve the data in the remote session we will use the DSI::datashield.assign.expr() function. This function runs a line of code on the remote session [^2], and in particular we want to run the function as.resource.object(), which is the DataSHIELD function in charge of resolving the resources.
[^2]: The same methodology and code can be used with unitary VCF resources that contain the variant information of all chromosomes.
lapply(1:21, function(x){ DSI::datashield.assign.expr(conns = conns, symbol = paste0("gds", x, "_object"), expr = as.symbol(paste0("as.resource.object(chr", x, ")"))) })
Now we have resolved the resources named chrXX into our remote R session. The objects retrieved have been assigned into variables named gdsXX_object. We can check the process was successful as we did before.
ds.class("gds1_object")
Assign the phenotypes
The objects we have loaded into our remote session are VCF files that contain genomic information of the individuals. To perform a GWAS this information has to be related to some phenotypes to extract relationships. Therefore, we need to load the phenotypes into the remote session. The phenotypes information is a table that contains the individuals as rows and phenotypes as columns. In this use case, we will use a resource (as with the VCF files) to load the phenotypes table into the remote session.
The procedure is practically the same as before with some minor tweaks. To begin, we have the phenotypes information in a single table (hence, a single resource). Then, instead of using the function as.resource.object, we will use as.resource.data.frame, this is because before we were loading a special object (VCF file) and now we are loading a plain table, so the internal treatment on the remote session has to be different.
DSI::datashield.assign.resource(conns, "pheno", "GWAS.ega_phenotypes") DSI::datashield.assign.expr(conns = conns, symbol = "pheno_object", expr = quote(as.resource.data.frame(pheno)))
We can follow the same analogy as before to know that we have assigned the phenotypes table to a variable called pheno_object on the remote R session.
ds.class("pheno_object")
We can also check the column names to see which information is present on the table.
ds.colnames("pheno_object")[[1]]
Merge VCF (genotype) and phenotype information {#vcf_merge}
Arrived at this point, we have 21 VCF objects (each one corresponds to a chromosome) and a phenotypes table on our remote session. The next step is merging each of the VCF objects with the phenotypes table. Before doing that however, we have to gather some information from the phenotypes table. The information to gather is summarized on the Table \@ref(tab:tab2).
+---------------------------------------------------------------+----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------+ | Information | Details | +===============================================================+====================================================================================================================================================================================================================================================+ | Which column has the samples identifier? | Column name that contains the IDs of the samples. Those are the IDs that will be matched to the ones present on the VCF objects. | +---------------------------------------------------------------+----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------+ | Which is the sex column on the covariates file? | Column name that contains the sex information. | +---------------------------------------------------------------+----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------+ | How are males and how are females encoded into this column? | There is not a standard way to encode the sex information, some people use 0/1; male/female; etc. Our approach uses a library that requires a specific encoding, for that reason we need to know the original encoding to perform the translation. | +---------------------------------------------------------------+----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------+ | Which is the case-control column? | Column name that contains the case/control to be studied. | +---------------------------------------------------------------+----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------+ | What is the encoding for case and for control on this column? | Similar as the sex column, the case/control will be translated to the particular standard of the software we have used to develop our functionalities [^3] . | +---------------------------------------------------------------+----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------+
: (#tab:tab2) Information to gather from the phenotypes table.
[^3]: It's important to note that all the other encodings present on the case-control column that are not the case or control, will be turned into missings. As an example, if we have "cancer"/"no cancer"/"no information"/"prefer not to answer" we can specifcy case="cancer", control="no cancer" and all the individuals with "no information"/"prefer not to answer" will be interpreted as missings.
If we are not sure about the exact column names, we can use the function ds.colnames as we did before. Also, we can use the function ds.table1D to check the level names of the categorical variables.
ds.table1D("pheno_object$sex")$counts ds.table1D("pheno_object$diabetes_diagnosed_doctor")$counts
From the different functions we used, we can extract our answers (Summarized on the Table \@ref(tab:tab3))
+-----------------------------------------------------------------+-----------------------------+ | Question | Answer | +=================================================================+=============================+ | Which column has the samples identifier? | "subject_id" | +-----------------------------------------------------------------+-----------------------------+ | Which is the sex column on the covariates file? | "sex" | +-----------------------------------------------------------------+-----------------------------+ | How are males and how are females encoded into this column? | "male"/"female" | +-----------------------------------------------------------------+-----------------------------+ | Which is the case-control column of interest of the covariates? | "diabetes_diagnosed_doctor" | +-----------------------------------------------------------------+-----------------------------+ | What is the encoding for case and for control on this column? | "Yes"/"No" | +-----------------------------------------------------------------+-----------------------------+
: (#tab:tab3) Summary of options from the example phenotypes table.
With all this information we can now merge the phenotypes and VCF objects into a type of object named GenotypeData. We will use the ds.GenotypeData function.
lapply(1:21, function(x){ ds.GenotypeData(x=paste0('gds', x,'_object'), covars = 'pheno_object', columnId = "subject_id", sexId = "sex", male_encoding = "male", female_encoding = "female", case_control_column = "diabetes_diagnosed_doctor", case = "Yes", control = "No", newobj.name = paste0('gds.Data', x), datasources = conns) })
The objects we just created are named gds.DataXX on the remote session. Now we are ready to perform a GWAS analysis. To perform a GWAS we have to supply this "Genotype Data" objects and some sort of formula in which we will specify the type of association we are interested on studying. The R language has it's own way of writing formulas, a simple example would be the following (relate the condition to the smoke variable adjusted by sex):
condition ~ smoke + sex
For this use case we will use the following formula.
diabetes_diagnosed_doctor ~ sex + hdl_cholesterol
Performing the GWAS
We are finally ready to achieve our ultimate goal, performing a GWAS analysis. We have our data (genotype + phenotypes) organized into the correspondent objects (GenotypeData) and our association formula ready. The function ds.metaGWAS is in charge of doing the analysis, we have to supply it with the names of the GenotypeData objects of the remote R session and the formula. Since we have 21 different objects, the paste0 function is used to simplify the call.
results_single <- ds.metaGWAS(genoData = paste0("gds.Data", 1:21), model = diabetes_diagnosed_doctor ~ sex + hdl_cholesterol)[[1]] results_single
We can display the results of the GWAS using a Manhattan plot.
manhattan(results_single)
Moreover, we can visualize the region with the lowest p-value in detail using the function LocusZoom. There are multiple arguments to control the gene annotation, display a different region of the GWAS, display a different range of positions and more. Make sure to check ?LocusZoom for all the details.
LocusZoom(results_single, use_biomaRt = FALSE)
datashield.logout(conns)
Multi cohorts
Now all the individuals used for the single cohort use case will be divided into three different synthetic cohorts. The results from each cohort will then be meta-analyzed to be compared to the results obtained by studying all the individuals together. As with the single-cohort methodology illustrated on the prior section, this method guarantees GDPR data confidentiality. The structure for a three cohort study is illustrated on the following figure (note this can be extrapolated for cohorts with a bigger (or lower) partner count).
knitr::include_graphics("gwas_images/multi_cohort.png")
The data analyst corresponds to the "RStudio" session, which through DataSHIELD Interface (DSI) connects with the Opal server located at the cohort network. This Opal server contains an array of resources that correspond to the different VCF files (sliced by chromosome) [^4] and a resource that corresponds to the phenotypes table of the studied individuals.
[^4]: The same methodology and code can be used with unitary VCF resources that contain the variant information of all chromosomes.
This use case will portray a complete study methodology, more precisely we will follow this flowchart:
knitr::include_graphics("gwas_images/multi_cohort_flowchart.png")
Brief description of each step:
- Step 1: Logging into the Opal servers and load all the VCF files to the remote R sessions. Corresponds to the sections Connection to the Opal server, Assign the VCF resources, Assign the phenotypes and Merge VCF (genotype) and phenotype information.
- Step 2: Performing a GWAS analysis on each cohort and collect all the aggregated non-disclosive statistics. Corresponds to the sections Performing the GWAS and [GWAS Plots].
- Step 3: Apply different QC control methodologies to assess possible problems on the data used on each cohort to perform the analysis. Corresponds to the section [Meta-level quality control].
- Step 4: Perform the meta-analysis of the aggregated statistics. Corresponds to the section Meta-analysis.
Connection to the Opal server
We have to create an Opal connection object to the different cohorts server. We do that using the following functions.
require('DSI') require('DSOpal') require('dsBaseClient') require('dsOmicsClient') builder <- DSI::newDSLoginBuilder() builder$append(server = "cohort1", url = "https://opal-demo.obiba.org/", user = "dsuser", password = "password", driver = "OpalDriver", profile = "omics") builder$append(server = "cohort2", url = "https://opal-demo.obiba.org/", user = "dsuser", password = "password", driver = "OpalDriver", profile = "omics") builder$append(server = "cohort3", url = "https://opal-demo.obiba.org/", user = "dsuser", password = "password", driver = "OpalDriver", profile = "omics") logindata <- builder$build() conns <- DSI::datashield.login(logins = logindata)
It is important to note that in this use case, we are only using one server that contains all the resources (https://opal-demo.obiba.org/), on this server there are all the resources that correspond to the different cohorts.
Assign the VCF resources {#multi-vcf}
Now that we have created a connection object to the different Opals, we have started three R session, our analysis will take place on those remote sessions, so we have to load the data into them.
In this use case we will use 63 different resources from the GWAS project hosted on the demo Opal server. This resources correspond to VCF files with information on individual chromosomes (21 chromosomes per three cohorts). The names of the resources are chrXXY (where XX is the chromosome number and Y is the cohort A/B/C). Following the Opal syntax, we will refer to them using the string GWAS.chrXXY.
We have to refer specifically to each different server by using conns[X], this allows us to communicate with the server of interest to indicate to it the resources that it has to load.
# Cohort 1 resources lapply(1:21, function(x){ DSI::datashield.assign.resource(conns[1], paste0("chr", x), paste0("GWAS.chr", x,"A")) }) # Cohort 2 resources lapply(1:21, function(x){ DSI::datashield.assign.resource(conns[2], paste0("chr", x), paste0("GWAS.chr", x,"B")) }) # Cohort 3 resources lapply(1:21, function(x){ DSI::datashield.assign.resource(conns[3], paste0("chr", x), paste0("GWAS.chr", x,"C")) })
Now we have assigned all the resources named GWAS.chrXXY into our remote R session. We have assigned them to the variables called chrXX. To verify this step has been performed correctly, we could use the ds.class function to check for their class and that they exist on the remote sessions.
ds.class("chr1")
We can see that the object chr1 exists in all the three servers.
Finally the resources are resolved to retrieve the data in the remote sessions.
lapply(1:21, function(x){ DSI::datashield.assign.expr(conns = conns, symbol = paste0("gds", x, "_object"), expr = as.symbol(paste0("as.resource.object(chr", x, ")"))) })
Now we have resolved the resources named chrXX into our remote R sessions. The objects retrieved have been assigned into variables named gdsXX_object. We can check the process was successful as we did before.
ds.class("gds1_object")
Assign the phenotypes {#assign-the-phenotypes-1}
The objects we have loaded into our remote sessions are VCF files that contain genomic information of the individuals. To perform a GWAS this information has to be related to some phenotypes to extract relationships. Therefore, we need to load the phenotypes into the remote sessions. The phenotypes information is a table that contains the individuals as rows and phenotypes as columns. In this use case, we will use a resource (as with the VCF files) to load the phenotypes table into the remote sessions.
As with the VCF resources, here we load a different resource to each cohort, as it is expected that each cohort has only the phenotypes information regarding their individuals, therefore different tables. Note that we are assigning the resource to the same variable at each server (pheno), so we only need one function call to resolve all the resources together.
# Cohort 1 phenotypes table DSI::datashield.assign.resource(conns[1], "pheno", "GWAS.ega_phenotypes_1") # Cohort 2 phenotypes table DSI::datashield.assign.resource(conns[2], "pheno", "GWAS.ega_phenotypes_2") # Cohort 3 phenotypes table DSI::datashield.assign.resource(conns[3], "pheno", "GWAS.ega_phenotypes_3") # Resolve phenotypes table DSI::datashield.assign.expr(conns = conns, symbol = "pheno_object", expr = quote(as.resource.data.frame(pheno)))
We can follow the same analogy as before to know that we have assigned the phenotypes table to a variable called pheno_object on the remote R session.
ds.class("pheno_object")
We can also check the column names to see which information is present on the table.
ds.colnames("pheno_object")[[1]]
Merge VCF (genotype) and phenotype information {#merge-vcf-genotype-and-phenotype-information}
Arrived at this point, we have 21 VCF objects at each cohort R session (each one corresponds to a chromosome) and a phenotypes table. The next step is merging each of the VCF objects with the phenotypes table. The same procedure as the single cohort can be applied to extract the arguments required.
With all this information we can now merge the phenotypes and VCF objects into a type of object named GenotypeData. We will use the ds.GenotypeData function.
lapply(1:21, function(x){ ds.GenotypeData(x=paste0('gds', x,'_object'), covars = 'pheno_object', columnId = "subject_id", sexId = "sex", male_encoding = "male", female_encoding = "female", case_control_column = "diabetes_diagnosed_doctor", case = "Yes", control = "No", newobj.name = paste0('gds.Data', x), datasources = conns) })
The objects we just created are named gds.DataXX on the remote session. Now we are ready to perform a GWAS analysis. We will use the same formula as the single cohort to be able to compare the results later:
diabetes_diagnosed_doctor ~ sex + hdl_cholesterol
Performing a meta - GWAS {#performing-the-gwas-1 .tabset .tabset-fade .tabset-pills}
We now proceed to perform the GWAS analysis.
results <- ds.metaGWAS(genoData = paste0("gds.Data", 1:21), model = diabetes_diagnosed_doctor ~ sex + hdl_cholesterol)
And we can see the results for each cohort.
Cohort 1 {.unnumbered}
results[[1]]
Cohort 2 {.unnumbered}
results[[2]]
Cohort 3 {.unnumbered}
results[[3]]
GWAS Plots {.tabset .tabset-fade .tabset-pills}
We can display the results of the GWAS using a Manhattan plot for each cohort.
Cohort 1 {.unnumbered}
manhattan(results[[1]])
Cohort 2 {.unnumbered}
manhattan(results[[2]])
Cohort 3 {.unnumbered}
manhattan(results[[3]])
Meta-level quality control
Up to this point we have collected the aggregated statistics from each cohort and we have visualized them. Before meta-analyzing this statistics we have to perform some quality control to make sure there are no major issues. There are three different methods bundled in dsOmicsClient to perform such task:
- EAF Plot: Plotting the reported EAFs against a reference set, such as from the HapMap or 1000 Genomes projects, or from one specific study, can help visualize patterns that pinpoint strand issues, allele miscoding or the inclusion of individuals whose self-reported ancestry did not match their genetic ancestry.
- P-Z Plot: Plot to reveal issues with beta estimates, standard errors and P values. The plots compare P values reported in the association GWAS with P values calculated from Z-statistics (P.ztest) derived from the reported beta and standard error. A study with no issues should show perfect concordance. It will generate a plot per study, where concordance of all the study SNPs will be displayed.
- SE-N Plot: Plot to reveal issues with trait transformations. A collection of studies with no issues will fall on a line.
If any of these QC raises some inconsistencies, please contact the correspondent study analyst.
EAF Plot
For this plot we are comparing the estimated allele frequencies to a reference set, so the first step is to get ourselves a reference set. For this example we will be using the ALL.wgs.phase1_release_v3.20101123 reference set. We will download it and load it into the client R session.
reference <- readr::read_tsv("https://homepages.uni-regensburg.de/~wit59712/easyqc/exomechip/ALL.wgs.phase1_release_v3.20101123.snps_indels_sv.sites.EUR_AF.txt.exm.tab.sexChr.DI.txt.GZ") reference
Now we just use the function eafPlot and we pass to it the GWAS results and the reference set.
eafPlot(x = results, reference = reference, rs_reference = "Rs", freq_reference = "Freq")
The obtained results are the expected, which is a linear correlation of slope one. This indicates the alleles are correctly encoded. For further explanation on how to assess the results produced by this plot, please refer to the Figure 4 of @winkler2014quality.
P-Z Plot
This plot will assess the issues with beta estimates, standard errors and P values. Here we will only need the results from the ds.GWAS function and pass them to the function pzPlot.
pzPlot(results)
Please note that all the SNPs are plotted, for that reason there may be some performance issues if there are many SNPs to be plotted.
A dataset with no issues will show perfect concordance. For further details please refer to the Figure 3 of @winkler2014quality.
SE-N Plot
Finally, we will assess issues with trait transformations. For this we will only need the results from the ds.GWAS function and pass them to the function seNPlot.
seNPlot(results)
If all the study points fall on a line, no issues are present. For further detail please refer to the Figure 2 of @winkler2014quality.
Meta-analysis
Given no problems have been detected on the meta-level QC, we will proceed. Up to this point, we have obtained association results for each cohort on the study. The next step is to combine this information using meta-analysis methods to derive a pooled estimate closest to the common truth. Each researcher might have an already built pipeline to do so, or a preferred method; nevertheless, we included a couple methods inside dsOmicsClient. They are the following:
- Meta-analysis of p-values: Using the sum of logs method (Fisher's method).
- Meta-analysis of beta values: Using a fixed-effects model. Methodology extracted and adapted from @betameta.
Assessment of the meta-analysis methods
Previously, we have calculated the same GWAS analysis both using all the individuals and a simulated three-cohort scenario. This gives us the ability to assess how good or bad are the different meta-analysis methods on discovering the closest common truth; to do so, we will compare both meta-analysis to the common truth, the evaluation method will be counting the number of top hits that match the common truth, more specifically we will search for matches on the top twenty hits.
We apply both methods to the multi-cohort use case results:
meta_pvalues <- metaPvalues(results) meta_bvalues <- metaBetaValues(results)
We can now compare the 20 top hits from the common truth (previously stored on the variable results_single) to the 20 top hits yielded by both meta-analysis methodologies.
# Extract 20 top hits for all three cases # All individuals top_20_complete <- results_single %>% arrange(p.value) %>% head(20) # Pvalue meta-analysis top_20_pval <- meta_pvalues %>% arrange(p.meta) %>% head(20) # Beta meta-analysis top_20_beta <- meta_bvalues %>% arrange(pval) %>% head(20) # Number of hits shared by all individuals case and beta meta-analysis sum(top_20_complete$rs %in% top_20_beta$rs) # Number of hits shared by all individuals case and pvalue meta-analysis sum(top_20_complete$rs %in% top_20_pval$rs)
The meta-analysis using beta values recovers a higher amount of top SNPs, therefore it can be considerated a more powerful method for GWAS meta-analyses.
Visualizing the results of the meta-analysis
We can use the manhattan plot and locus zoom to visualize the results of the meta-analysis as we visualized the single cohorts before. To help assess the meta-analysis behaviour, we will plot together the meta-analysis results (left plot) and the common truth results (right plot).
Manhatan plot {.tabset .tabset-fade .tabset-pills}
Meta-analysis {.unnumbered}
manhattan(meta_bvalues)
Common truth {.unnumbered}
manhattan(results_single)
LocusZoom {.tabset .tabset-fade .tabset-pills}
Meta-analysis {#meta-analysis-2 .unnumbered}
LocusZoom(meta_bvalues, pvalue = "p.F", use_biomaRt = FALSE)
Common truth {.unnumbered}
LocusZoom(results_single, use_biomaRt = FALSE)
Meta-analysis versus pooled linear models
The dsOmicsClient package also bundles a function to fit pooled generalized linear models (GLM) on single (or multiple) SNPs. On this section we will compare the results of the meta-analysis to the pooled GLM models to see which method is closer to the common truth. The meta-analysis method will be the beta values (fixed-effects model).
The study methodology will be similar to the one we just performed. The top 20 hits yielded by the meta-analysis will be selected, those SNPs will be studied using the pooled GLM function. Both results will be compared to the common truth.
We aim to replicate a real scenario with this study, typically there is no access to the common truth (if so there is no point on performing meta-analysis on synthetic multi-cohorts), therefore we simulate the discovery of SNPs of interest via meta-analysis, then we want to assess if studying individually those SNPs using pooled GLM will put us closer to the common truth than what we already achieved.
We have already calculated the top 20 SNPs discovered by the beta meta-analysis.
top_20_beta
Next, we have to fit pooled GLM using the ds.glmSNP function, obviously we have to fit the same model that we did on the GWAS (Diabetes.diagnosed.by.doctor ~ sex + HDL.cholesterol), otherwise the results will not be consistent. Remember that we have the genotype data separated by chromosome, following that we will construct the GLM calls as follows:
s <- lapply(1:nrow(top_20_beta), function(x){ tryCatch({ ds.glmSNP(snps.fit = top_20_beta[x, 1], model = diabetes_diagnosed_doctor ~ sex + hdl_cholesterol, genoData = paste0("gds.Data", top_20_beta[x, 2])) }, error = function(w){NULL}) }) # Simple data cleaning and column name changes ss <- data.frame(do.call(rbind, s)) ss <- ss %>% rownames_to_column("rs") pooled <- ss %>% dplyr::rename(Est.pooled = Estimate, SE.pooled = Std..Error, pval.pooled = p.value) %>% dplyr::select(-c(n, p.adj))
# ds.glmSNP(snps.fit = unlist(top_20_beta[3:5, 1]), model = diabetes_diagnosed_doctor ~ sex + hdl_cholesterol, genoData = paste0("gds.Data", unlist(top_20_beta[3:5, 2])), mc.cores = 3)
We now extract the results of interest from the meta-analysis and the common truth.
# Get data from all individuals study original <- results_single %>% dplyr::rename(pval.original = p.value, Est.original = Est, SE.original = Est.SE) %>% dplyr::select(c(rs, Est.original, SE.original, pval.original)) # Arrange dadta from beta meta-analysis beta <- top_20_beta %>% dplyr::rename(pval.beta = pval, Est.beta = b.F, SE.beta = se.F) %>% dplyr::select(c(rs, Est.beta, SE.beta, pval.beta)) # Merge all data on a single table total <- Reduce(function(x,y){inner_join(x,y, by = "rs")}, list(original, pooled, beta))
With all the information gathered we can visualize the results on the Table 4.
[Table 4: Beta values, standard errors and p-values yielded by the three methods: GWAS of all individuals, meta-analysis of synthetic cohorts, pooled analysis of synthetic cohorts.]{#tab:tab4 style="font-size:8pt"}
library(gt) total %>% gt() %>% tab_spanner(label = 'Beta', columns = c(Est.original, Est.pooled, Est.beta)) %>% tab_spanner(label = 'SE', columns = c(SE.original, SE.pooled, SE.beta)) %>% tab_spanner(label = 'P-Value', columns = c(pval.original, pval.pooled, pval.beta)) %>% cols_label(Est.original = "Original", Est.pooled = "Pooled", Est.beta = "Meta", SE.original = "Original", SE.pooled = "Pooled", SE.beta = "Meta", pval.original = "Original", pval.pooled = "Pooled", pval.beta = "Meta", rs = "SNP id") %>% fmt_number(columns = c(Est.original, Est.pooled, Est.beta, SE.original, SE.pooled, SE.beta), decimals = 3) %>% fmt_scientific(columns = c(pval.original, pval.pooled, pval.beta), decimals = 1) %>% tab_options(column_labels.font.weight = "bold") %>% tab_style(style = cell_fill(color = "grey"), locations = list(cells_body(columns = c(SE.original, SE.pooled, SE.beta), rows = c(seq(2,nrow(total),2))))) %>% tab_style(style = cell_fill(color = "lightgray"), locations = list(cells_body(columns = c(SE.original, SE.pooled, SE.beta), rows = c(seq(1,nrow(total),2))))) %>% tab_style(style = cell_borders(sides = "all", color = "lightgrey", style = "solid", weight = px(1)), locations = cells_body(columns = everything(), rows = everything())) %>% tab_style(style = cell_text(color = "red"), locations = list(cells_body(columns = Est.original, rows = Est.original<0))) %>% tab_style(style = cell_text(color = "red"), locations = list(cells_body(columns = Est.beta, rows = Est.beta<0))) %>% tab_style(style = cell_text(color = "red"), locations = list(cells_body(columns = Est.pooled, rows = Est.pooled<0))) %>% tab_options(table.font.size = "10pt")
Finally, to conclude this study we can compute the bias and mean squared error (code not shown). The results are shown on the Table 5.
# MSE mse_est_pooled <- mean((total$Est.original - total$Est.pooled)^2) mse_est_beta <- mean((total$Est.original - total$Est.beta)^2)
# Bias bias_est_pooled <- mean(total$Est.original - total$Est.pooled) bias_est_beta <- mean(total$Est.original - total$Est.beta)
statistics <- data.frame(MSE = c(mse_est_pooled, mse_est_beta), Bias = c(bias_est_pooled, bias_est_beta), stat = c("Beta", "Beta"), var = c("Pooled", "Meta"))
[Table 5: Mean squared error and bias for the betas from the meta-analysis and pooled analysis compared to the common truth (all individuals).]{#tab:tab5 style="font-size:8pt"}
statistics %>% group_by(stat) %>% gt(rowname_col = "var") %>% fmt_scientific(columns = c(MSE, Bias), decimals = 1) %>% tab_options(column_labels.font.weight = "bold") %>% tab_options(table.font.size = "10pt")
With this results we can conclude that refining the statistics for the SNPs of interest using pooled GLM is advisable. Therefore the recommended flowchart when performing a GWAS using dsOmicsClient+DataSHIELD is the following:
REVISAR ESTOS RESULTADOS
- Perform GWAS with
dsOmicsClient::dsGWAS - Perform meta-analysis on the results with
dsOmicsClient::metaBetaValues - Extract top hits from the meta-analysis.
- Perform pooled GLM on top hits with
dsOmicsClient::ds.glmSNP
Fast GWAS analysis
An alternate methodology to performing a meta analysis is to use the fast GWAS methodology. This method is named after the performance it achieves, however, it does provide further advantages other than computational speed. The main advantage is that it does not compute the results for each cohort to then be meta-analyzed, it actually computes them using a pooled technique that achieves the exact same results when all the individuals are together than when they are separated by cohorts. This is of great importance as there is no need to worry about cohort-imbalance, which is usually a problem in multi-cohort meta-analysis studies. The only downside of this method is that it does not yield the same results of a traditional GWAS analysis pipeline, the inacuraccy of the results however is really low, detailed information about that can be found on the paper that proposes the method.
The usage is similar to the meta-GWAS with a couple of added options, feel free to check the documentation with ?ds.fastGWAS to see information about them.
results_fast <- ds.fastGWAS(genoData = paste0("gds.Data", 1:21), formula = "diabetes_diagnosed_doctor ~ sex + hdl_cholesterol", family = "binomial", snpBlock = 5000)
datashield.logout(conns)
GWAS Plot
We can display the results of the fast GWAS using a Manhattan plot.
manhattan(results_fast, pvalCol = 4)
Meta-analysis versus fast GWAS
To finish the GWAS section we can evaluate the results of the beta meta-analysis and the results of the fast GWAS. Both methods will be compared to the ground truth.
As before we can start by seeing the amount of SNPs that both methodologies recover as top hits.
# Extract 20 top hits for all three cases # All individuals top_20_complete <- results_single %>% arrange(p.value) %>% head(20) # Fast GWAS top_20_fast <- results_fast %>% arrange(p.value) %>% head(20) # Beta meta-analysis top_20_beta <- meta_bvalues %>% arrange(pval) %>% head(20) # Number of hits shared by all individuals case and beta meta-analysis sum(top_20_complete$rs %in% top_20_beta$rs) # Number of hits shared by all individuals case and fast GWAS sum(top_20_complete$rs %in% top_20_fast$rs)
On that regard it seems the meta-analysis is performing better.
We now extract the results of interest from the meta-analysis and the common truth.
# Get data from all individuals study original <- results_single %>% dplyr::rename(pval.original = p.value, Est.original = Est, SE.original = Est.SE) %>% dplyr::select(c(rs, Est.original, SE.original, pval.original)) # Arrange dadta from beta meta-analysis beta <- top_20_beta %>% dplyr::rename(pval.beta = pval, Est.beta = b.F, SE.beta = se.F) %>% dplyr::select(c(rs, Est.beta, SE.beta, pval.beta)) # Arrange data from fast GWAS fast <- top_20_fast %>% dplyr::rename(pval.fast = p.value, Est.fast = Est, SE.fast = Est.SE) %>% dplyr::select(c(rs, Est.fast, SE.fast, pval.fast)) # Merge all data on a single table total <- Reduce(function(x,y){inner_join(x,y, by = "rs")}, list(original, fast, beta))
With all the information gathered we can visualize the results on the Table 6.
[Table 6: Beta values, standard errors and p-values yielded by the three methods: GWAS of all individuals, meta-analysis of synthetic cohorts, fast GWAS.]{#tab:tab6 style="font-size:8pt"}
library(gt) total %>% gt() %>% tab_spanner(label = 'Beta', columns = c(Est.original, Est.fast, Est.beta)) %>% tab_spanner(label = 'SE', columns = c(SE.original, SE.fast, SE.beta)) %>% tab_spanner(label = 'P-Value', columns = c(pval.original, pval.fast, pval.beta)) %>% cols_label(Est.original = "Original", Est.fast = "Fast", Est.beta = "Meta", SE.original = "Original", SE.fast = "Fast", SE.beta = "Meta", pval.original = "Original", pval.fast = "Fast", pval.beta = "Meta", rs = "SNP id") %>% fmt_number(columns = c(Est.original, Est.fast, Est.beta, SE.original, SE.fast, SE.beta), decimals = 3) %>% fmt_scientific(columns = c(pval.original, pval.fast, pval.beta), decimals = 1) %>% tab_options(column_labels.font.weight = "bold") %>% tab_style(style = cell_fill(color = "grey"), locations = list(cells_body(columns = c(SE.original, SE.fast, SE.beta), rows = c(seq(2,nrow(total),2))))) %>% tab_style(style = cell_fill(color = "lightgray"), locations = list(cells_body(columns = c(SE.original, SE.fast, SE.beta), rows = c(seq(1,nrow(total),2))))) %>% tab_style(style = cell_borders(sides = "all", color = "lightgrey", style = "solid", weight = px(1)), locations = cells_body(columns = everything(), rows = everything())) %>% tab_style(style = cell_text(color = "red"), locations = list(cells_body(columns = Est.original, rows = Est.original<0))) %>% tab_style(style = cell_text(color = "red"), locations = list(cells_body(columns = Est.beta, rows = Est.beta<0))) %>% tab_style(style = cell_text(color = "red"), locations = list(cells_body(columns = Est.fast, rows = Est.fast<0))) %>% tab_options(table.font.size = "10pt")
Finally, to conclude this study we can compute the bias and mean squared error (code not shown). The results are shown on the Table 7.
# MSE mse_est_fast <- mean((total$Est.original - total$Est.fast)^2) mse_est_beta <- mean((total$Est.original - total$Est.beta)^2)
# Bias bias_est_fast <- mean(total$Est.original - total$Est.fast) bias_est_beta <- mean(total$Est.original - total$Est.beta)
statistics <- data.frame(MSE = c(mse_est_fast, mse_est_beta), Bias = c(bias_est_fast, bias_est_beta), stat = c("Beta", "Beta"), var = c("Fast", "Meta"))
[Table 7: Mean squared error and bias for the betas from the meta-analysis and fast GWAS compared to the common truth (all individuals).]{#tab:tab5 style="font-size:8pt"}
statistics %>% group_by(stat) %>% gt(rowname_col = "var") %>% fmt_scientific(columns = c(MSE, Bias), decimals = 1) %>% tab_options(column_labels.font.weight = "bold") %>% tab_options(table.font.size = "10pt")
On this brief comparison we've seen that the fast GWAS retrieves sensibly less SNPs from the ground truth but it has better performance of bias and MSE. It must be noted that the data used for this GWAS examples does not suffer from any cohort imbalance, so the meta-analysis is expected to yield proper results, on a different environment the fast GWAS is expected to exceed the performance of the meta-analysis in a more remarkable manner.
Post omic analysis
Up to this point we have analyzed the genotype and phenotype data by performing a genome wide association study. We can further study the yielded results by performing an enrichment analysis.
Enrichment analysis
The GWAS results are a collection of SNPs that displayed a relationship with a given phenotype. Those SNPs may fall inside gene regions, and those genes can be functionally related. Studying this can help researchers understand the underlying biological processes. Those analysis are carried by querying the selected gene to a database, for this example we will be using the DisGeNET database.
Annotation of genes
The first step of the enrichment analysis is to annotate the genes that contain the SNPs of interest yielded by the GWAS. There are many ways to perform this step, we propose to use the biomaRt package and retrieve the annotation from the ensembl genome browser.
First we select the SNPs of interest. We have established a p.value threshold to do so.
snps_of_interest <- results_fast %>% dplyr::arrange(p.value) %>% dplyr::select(chr, pos, rs, p.value) %>% dplyr::filter(p.value < 0.00005)
And with this list of SNPs we can move to the annotation.
library(biomaRt) # Perform ensembl connection gene.ensembl <- useEnsembl(biomart = "ensembl", dataset = "hsapiens_gene_ensembl", GRCh = 37) # Query SNP by SNP genes_interest <- Reduce(rbind, apply(snps_of_interest, 1, function(x){ result <- getBM( attributes = c('entrezgene_id', 'chromosome_name', 'ensembl_gene_id', 'start_position', "end_position"), filters = c('start', 'end', 'chromosome_name'), values = list(start = x[2], end = x[2], chromosome_name = x[1]), mart = gene.ensembl) }))
Enrichment analysis query
Having the genes of interest annotated, the final step is to query them to DisGeNET. We will use the function enrichDGN from the package DOSE for it. This package is part of a large ecosystem of packages dedicated to enrichment analysis, this allows for easy representation of the query results.
library(DOSE) enrichment <- enrichDGN(unlist(genes_interest$entrezgene_id))
Enrichment analysis plots
Here we will portray some of the visualizations that can be performed with the query results. For more information and further visualization options please read the official vignette.
Barplot
# library(enrichplot) # barplot(enrichment)
Dotplot
# dotplot(enrichment)
CNET plot
# library(ggnewscale) # # convert gene ID to Symbol # edox <- setReadable(enrichment, 'org.Hs.eg.db', 'ENTREZID') # cnetplot(edox, foldChange=genes_interest$entrezgene_id)
Polygenic risk scores
Definition
By checking for specific variants and quantifying them, a researcher can extract the polygenic risk score (PRS) of an individual, which translates to an associated risk of the individual versus a concrete phenotype. A definition of the term reads
"A weighted sum of the number of risk alleles carried by an individual, where the risk alleles and their weights are defined by the loci and their measured effects as detected by genome wide association studies." (Extracted from @torkamani2018personal)
The use of PRS markers is very popular nowadays and it has been proved to be a good tool to asses associated risks @escott2017polygenic, @forgetta2020development, @kramer2020breast.
When calculating the PRS using DataSHIELD, the actual scores are not returned, as it would not be GDPR compliant to be able to determine the risk of an individual versus a certain disease. The actual results are stored on the servers and optionally can be merged to the phenotypes table. This allows further study of the PRS using summaries or fitting pooled GLM models to assess relationships between phenotypes and PRS scores.
Study configuration
The PRS use case will be set up as a multi-cohort, using the same dataset as the multi-cohort GWAS (same VCF resources). A single cohort PRS could be performed analogously using the single cohort example of the GWAS.
knitr::include_graphics("gwas_images/prs.png")
Source of polygenic scores
There are two ways of stating the SNPs of interest and their weights in order to calculate the PRS.
-
Providing a
prs_table(SNPs of interest) table. Theprs_tabletable has to have a defined structure and column names in order to be understood by this function. This table can be formulated using two schemas:- Scheme 1: Provide SNP positions. Use the column names: "
chr_name", "chr_position", "effect_allele", "reference_allele", "effect_weight"
| chr_name | chr_position | effect_allele | reference_allele | effect_weight | |----------|--------------|---------------|------------------|---------------| | 1 | 100880328 | T | A | 0.0373 | | 1 | 121287994 | G | A | -0.0673 | | 6 | 152023191 | A | G | 0.0626 | | ... | ... | ... | ... | ... |
- Scheme 2: Provide SNP id's. Use the column names: "
rsID", "effect_allele", "reference_allele", "effect_weight".
| rdID | effect_allele | reference_allele | effect_weight | |------------|---------------|------------------|---------------| | rs11206510 | T | C | 0.0769 | | rs4773144 | G | A | 0.0676 | | rs17514846 | A | C | 0.0487 | | ... | ... | ... | ... |
- Scheme 1: Provide SNP positions. Use the column names: "
The effect_weight have to be the betas (log(OR)).
- Provide a PGS Catalog 'Polygenic Score ID & Name'. If this option is in use, the SNPs and beta weights will be downloaded from the PGS Catalog and will be used to calculate the PRS.
Please note when using a custom prs_table table that it is much faster to use the Schema 1, as the subset of the VCF files is miles faster to do using chromosome name and positions rather than SNP id's.
Connection to the Opal server {#connection-to-the-opal-server-2}
We have to create an Opal connection object to the cohort server. We do that using the following functions.
require('DSI') require('DSOpal') require('dsBaseClient') require('dsOmicsClient') builder <- DSI::newDSLoginBuilder() builder$append(server = "cohort1", url = "https://opal-demo.obiba.org/", user = "dsuser", password = "password", driver = "OpalDriver", profile = "omics") builder$append(server = "cohort2", url = "https://opal-demo.obiba.org/", user = "dsuser", password = "password", driver = "OpalDriver", profile = "omics") builder$append(server = "cohort3", url = "https://opal-demo.obiba.org/", user = "dsuser", password = "password", driver = "OpalDriver", profile = "omics") logindata <- builder$build() conns <- DSI::datashield.login(logins = logindata)
Assign the VCF resources
We assign the VCF and phenotypes resources.
# Cohort 1 resources lapply(1:21, function(x){ DSI::datashield.assign.resource(conns[1], paste0("chr", x), paste0("GWAS.chr", x,"A")) }) DSI::datashield.assign.resource(conns[1], "phenotypes_resource", "GWAS.ega_phenotypes_1") # Cohort 2 resources lapply(1:21, function(x){ DSI::datashield.assign.resource(conns[2], paste0("chr", x), paste0("GWAS.chr", x,"B")) }) DSI::datashield.assign.resource(conns[2], "phenotypes_resource", "GWAS.ega_phenotypes_2") # Cohort 3 resources lapply(1:21, function(x){ DSI::datashield.assign.resource(conns[3], paste0("chr", x), paste0("GWAS.chr", x,"C")) }) DSI::datashield.assign.resource(conns[3], "phenotypes_resource", "GWAS.ega_phenotypes_3")
And we resolve the phenotypes table. Remember to pass the table through the ds.make_valid_names function to be sure the column names have valid R syntax.
DSI::datashield.assign.expr(conns = conns, symbol = "phenotypes", expr = as.symbol("as.resource.data.frame(phenotypes_resource)")) ds.make_valid_names("phenotypes")
There is one crucial difference between this analysis and the GWAS, here we don't need to resolve the resources. This is for memory efficiency purposes. When we calculate a PRS, we are only interested in a small selection of SNPs, for that reason the (potentially) huge VCF files will be trimmed before being resolved, so only the relevant SNPs are loaded in the memory. For that reason the resolving of the resources is delegated to the ds.PRS function, which will perform this trimming according to the SNPs of interest yielded by the PGS catalog query or the prs_table table.
Calculate the PRS
The PRS results will be added to the phenotypes table to be later studied using a pooled GLM. To do that, the following arguments of the function ds.PRS will be used:
table: Name of the table where the results will be merged.table_id_column: Number of the column in thetabletable that contains the individuals ID to merge the results from the PRS.table_prs_name: Name to append to the PRS results. It will take the structureprs_table_prs_nameandprs_nw_table_prs_name.
PGS Catalog Example
For this example we will calculate the PRS associated to the trait 'HDL Cholesterol' and 'Thyroid cancer', which corresponds to the PGS ID PGS000660 and PGS000636
resources <- paste0("chr", 1:21) names(resources) <- paste0("chr", 1:21) resources <- as.list(resources) # HDL cholesterol ds.PRS(resources = resources, pgs_id = "PGS000660", table = "phenotypes", table_id_column = "subject_id") # Thyroid cancer ds.PRS(resources = resources, pgs_id = "PGS000636", table = "phenotypes", table_id_column = "subject_id")
We can check that the results have been correctly added to the phenotypes table.
tail(ds.colnames('phenotypes')[[1]])
Custom prs_table example
Here we will illustrate a use case with a custom prs_table, please note that the proposed SNPs of interest and betas are totally arbitrary.
We will first assemble the prs_table table following the indications found on the Details section of ?ds.PRS (Schema 1).
prs_table <- data.frame(chr_name = c(9, 9, 9, 9, 1, 1, 2, 2), chr_position = c(177034, 161674, 140859, 119681, 60351, 825410, 154629, 145710), effect_allele = c("T", "C", "G", "C", "G", "G", "T", "A"), reference_allele = c("G", "T", "A", "T", "A", "A", "G", "G"), effect_weight = c(0.5, 0.2, 0.1, 0.2, 0.1, 0.3, 0.1, 0.8))
Now we can pass the custom prs_table to the PRS function to retrieve the aggregated results.
resources <- paste0("chr", 1:21) names(resources) <- paste0("chr", 1:21) resources <- as.list(resources) ds.PRS(resources = resources, prs_table = prs_table, table = "phenotypes", table_id_column = "subject_id", table_prs_name = "custom_prs_table_results")
Study of the PRS results
We can fit pooled GLM models using the function ds.glm. In this use case we will fit a model to relate the HDL.cholesterol to prs_PGS000660 adjusted by sex. The same will be done with the prs_PGS000636.
ds.glm(formula = "hdl_cholesterol ~ prs_PGS000660 + sex", family = "gaussian", data = "phenotypes")$coefficients ds.glm(formula = "hdl_cholesterol ~ prs_PGS000636 + sex", family = "gaussian", data = "phenotypes")$coefficients
datashield.logout(conns)
Technical details
GWAS
TODO
Internal structure of the PRS functionality
The PRS function can be divided into three main blocks that perform the required tasks to come up with the results. The three functional blocks are the following:
- Client: Assemble the
prs_tabletable if PGS Catalog is used, perform calls to the server to subset the resources and calculate the PRS with the subsetted resources. - Server resolver: Resolve the selected resources (VCF files) and subset them using the prs_table.
- Server analysis: Use the resolved resources (small VCF files with only the SNPs of interest according to the
prs_tabletable) to calculate the PRS.
In order to introduce a little how those three blocks work internally, schematized flow charts have been designed. To understand the exact inner working of the functionality it is advised to follow the flowcharts alongside the actual source code. Find the links to the three blocks source code, client, server resolver and server analysis.
Client
The client performs two basic tasks. The first one is to select between a user introduced prs_table or to retrieve the prs_table from the PGS Catalog. Please note that prs_table stands for Region Of Interest, which may not be the common term for PGS analysis but it makes sense since it refers to a table that contain the SNP's of interest and their weight to the score. If the user introduces a custom prs_table and a PGS Catalog ID, only the introduced table will be used, discarding the PGS Catalog ID information. Once this table is assembled, the next step is to call the required functions on the study servers. First, the resource resolver is called, and after that, the function that calculates the PRS is called.
This is illustrated on the following figure.
knitr::include_graphics("gwas_images/block1.png")
Server resolver
The server resolver is in charge of resolving the resources. The interesting aspect is that only a region of interest is actually assigned to the R session, this is to avoid overloading the memory with unnecessary information. There are two different methodologies to perform this subsetting, one is using chromosome names and position and the other is using the SNP id's. Due to the technical specification of VCF files, is much easier to perform the subsetting using chromosome names and positions because there is an indexing file for the VCF files to perform fast queries of regions by position. On the other hand, to filter a VCF file using SNP id's, the whole file has to be scanned, yielding a much slower process.
This block is illustrated on the following figure.
knitr::include_graphics("gwas_images/block2.png")
Server analysis
Many processes are performed inside the analysis block. For that reason, more than a flow chart, a step by step guide has been illustrated with the objects that are created (or modified) on each step. The most important step on this block is making sure that the alternate (risk) alleles match between the VCF resources and the alternate alleles stated by the prs_table or PGS Catalog.
The block is illustrated on the following figure.
knitr::include_graphics("gwas_images/block3.png")
References
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
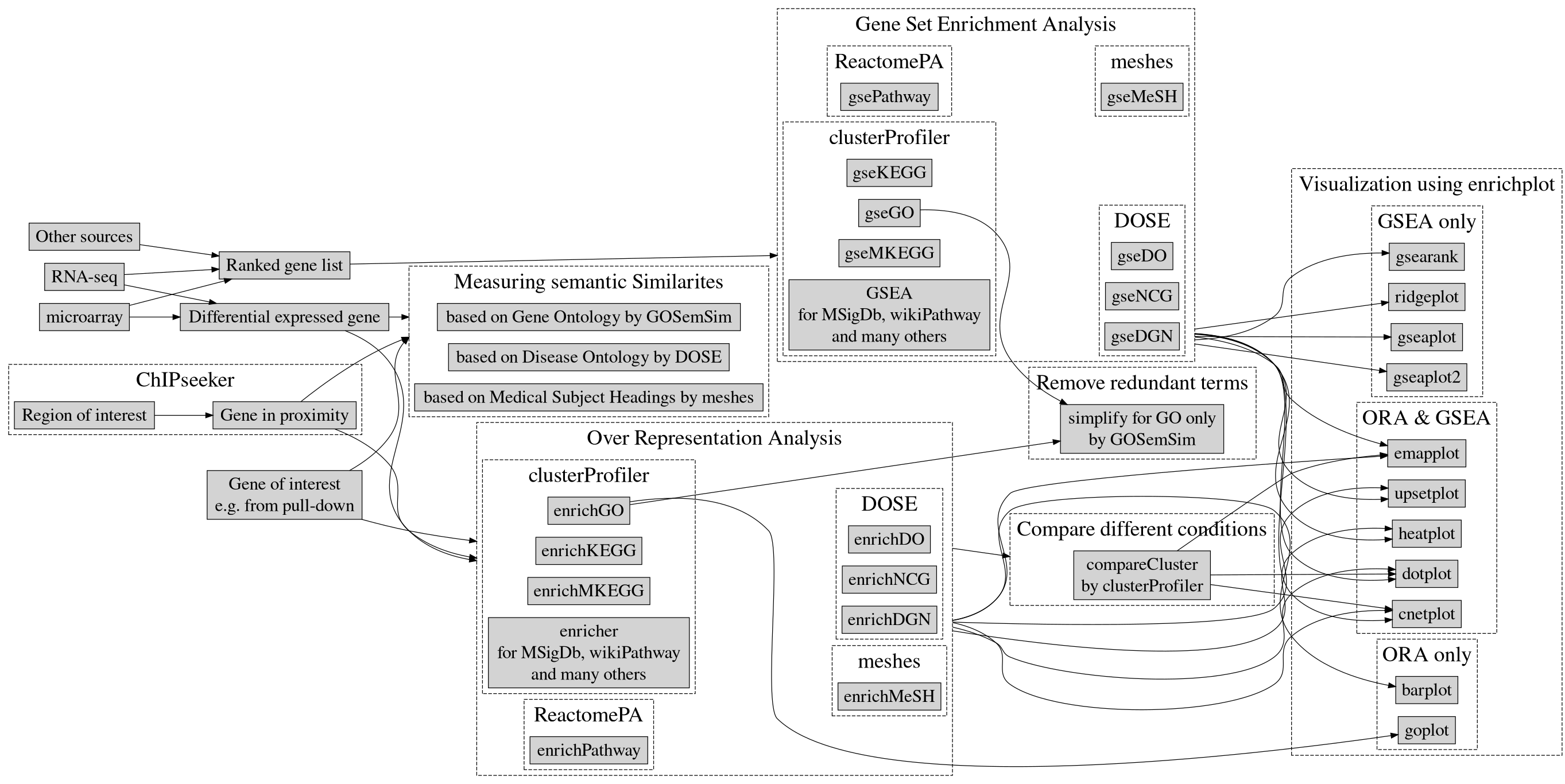{kind=link}
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.