In saezlab/decoupleR: decoupleR: Ensemble of computational methods to infer biological activities from omics data
knitr::opts_chunk$set(
collapse = TRUE,
comment = "#>"
)
Bulk RNA-seq yield many molecular readouts that are hard to interpret by
themselves. One way of summarizing this information is by inferring
transcription factor (TF) activities from prior knowledge.
In this notebook we showcase how to use decoupleR for transcription factor activity
inference with a bulk RNA-seq data-set where the transcription factor FOXA2 was
knocked out in pancreatic cancer cell lines.
The data consists of 3 Wild Type (WT) samples and 3 Knock Outs (KO). They are
freely available in
GEO.
Loading packages
First, we need to load the relevant packages:
## We load the required packages
library(decoupleR)
library(dplyr)
library(tibble)
library(tidyr)
library(ggplot2)
library(pheatmap)
library(ggrepel)
Loading the data-set
Here we used an already processed bulk RNA-seq data-set. We provide the
normalized log-transformed counts, the experimental design meta-data and the
Differential Expressed Genes (DEGs) obtained using limma.
For this example we use limma but we could have used DeSeq2, edgeR or any
other statistical framework. decoupleR requires a gene level statistic to
perform enrichment analysis but it is agnostic of how it was generated. However,
we do recommend to use statistics that include the direction of change and its
significance, for example the t-value obtained for limma(t) or DeSeq2(stat).
edgeR does not return such statistic but we can create our own by weighting the
obtained logFC by pvalue with this formula: -log10(pvalue) * logFC.
We can open the data like this:
inputs_dir <- system.file("extdata", package = "decoupleR")
data <- readRDS(file.path(inputs_dir, "bk_data.rds"))
From data we can extract the mentioned information. Here we see the normalized
log-transformed counts:
# Remove NAs and set row names
counts <- data$counts %>%
dplyr::mutate_if(~ any(is.na(.x)),
~ dplyr::if_else(is.na(.x), 0, .x)) %>%
tibble::column_to_rownames(var = "gene") %>%
as.matrix()
head(counts)
The design meta-data:
design <- data$design
design
And the results of limma, of which we are interested in extracting the
obtained t-value and p-value from the contrast:
# Extract t-values per gene
deg <- data$limma_ttop %>%
dplyr::select(ID, logFC, t, P.Value) %>%
dplyr::filter(!is.na(t)) %>%
tibble::column_to_rownames(var = "ID") %>%
as.matrix()
head(deg)
CollecTRI network
CollecTRI is a comprehensive resource
containing a curated collection of TFs and their transcriptional targets
compiled from 12 different resources. This collection provides an increased
coverage of transcription factors and a superior performance in identifying
perturbed TFs compared to our previous
DoRothEA network and other literature
based GRNs. Similar to DoRothEA, interactions are weighted by their mode of
regulation (activation or inhibition).
For this example we will use the human version (mouse and rat are also
available). We can use decoupleR to retrieve it from OmniPath. The argument
split_complexes keeps complexes or splits them into subunits, by default we
recommend to keep complexes together.
net <- decoupleR::get_collectri(organism = 'human',
split_complexes = FALSE)
net
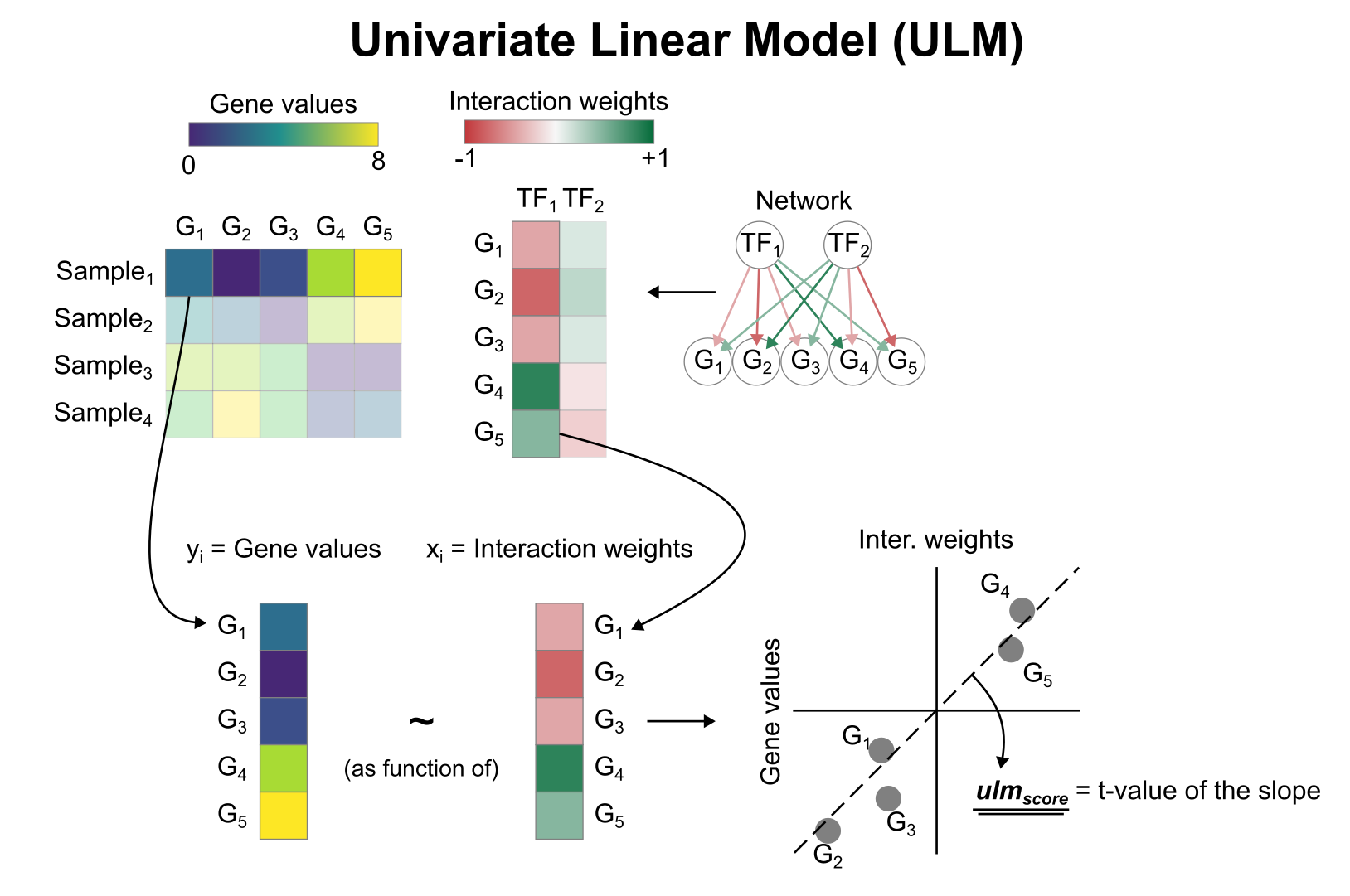
Activity inference with Univariate Linear Model (ULM)
To infer TF enrichment scores we will run the Univariate Linear Model (ulm) method. For each sample in our dataset (mat) and each TF in our network (net), it fits a linear model that predicts the observed gene expression
based solely on the TF's TF-Gene interaction weights. Once fitted, the obtained t-value of the slope is the score. If it is positive, we interpret that the TF is active and if it is negative we interpret that it is inactive.
To run decoupleR methods, we need an input matrix (mat), an input prior
knowledge network/resource (net), and the name of the columns of net that we
want to use.
# Run ulm
sample_acts <- decoupleR::run_ulm(mat = counts,
net = net,
.source = 'source',
.target = 'target',
.mor = 'mor',
minsize = 5)
sample_acts
Visualization
From the obtained results we will observe the most variable activities across samples in a heat-map:
n_tfs <- 25
# Transform to wide matrix
sample_acts_mat <- sample_acts %>%
tidyr::pivot_wider(id_cols = 'condition',
names_from = 'source',
values_from = 'score') %>%
tibble::column_to_rownames('condition') %>%
as.matrix()
# Get top tfs with more variable means across clusters
tfs <- sample_acts %>%
dplyr::group_by(source) %>%
dplyr::summarise(std = stats::sd(score)) %>%
dplyr::arrange(-abs(std)) %>%
head(n_tfs) %>%
dplyr::pull(source)
sample_acts_mat <- sample_acts_mat[,tfs]
# Scale per sample
sample_acts_mat <- scale(sample_acts_mat)
# Choose color palette
colors <- rev(RColorBrewer::brewer.pal(n = 11, name = "RdBu"))
colors.use <- grDevices::colorRampPalette(colors = colors)(100)
my_breaks <- c(seq(-2, 0, length.out = ceiling(100 / 2) + 1),
seq(0.05, 2, length.out = floor(100 / 2)))
# Plot
pheatmap::pheatmap(mat = sample_acts_mat,
color = colors.use,
border_color = "white",
breaks = my_breaks,
cellwidth = 15,
cellheight = 15,
treeheight_row = 20,
treeheight_col = 20)
We can also infer TF activities from the t-values of the DEGs between KO
and WT:
# Run ulm
contrast_acts <- decoupleR::run_ulm(mat = deg[, 't', drop = FALSE],
net = net,
.source = 'source',
.target = 'target',
.mor='mor',
minsize = 5)
contrast_acts
Let's show the changes
in activity between KO and WT:
# Filter top TFs in both signs
f_contrast_acts <- contrast_acts %>%
dplyr::mutate(rnk = NA)
msk <- f_contrast_acts$score > 0
f_contrast_acts[msk, 'rnk'] <- rank(-f_contrast_acts[msk, 'score'])
f_contrast_acts[!msk, 'rnk'] <- rank(-abs(f_contrast_acts[!msk, 'score']))
tfs <- f_contrast_acts %>%
dplyr::arrange(rnk) %>%
head(n_tfs) %>%
dplyr::pull(source)
f_contrast_acts <- f_contrast_acts %>%
filter(source %in% tfs)
colors <- rev(RColorBrewer::brewer.pal(n = 11, name = "RdBu")[c(2, 10)])
p <- ggplot2::ggplot(data = f_contrast_acts,
mapping = ggplot2::aes(x = stats::reorder(source, score),
y = score)) +
ggplot2::geom_bar(mapping = ggplot2::aes(fill = score),
color = "black",
stat = "identity") +
ggplot2::scale_fill_gradient2(low = colors[1],
mid = "whitesmoke",
high = colors[2],
midpoint = 0) +
ggplot2::theme_minimal() +
ggplot2::theme(axis.title = element_text(face = "bold", size = 12),
axis.text.x = ggplot2::element_text(angle = 45,
hjust = 1,
size = 10,
face = "bold"),
axis.text.y = ggplot2::element_text(size = 10,
face = "bold"),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank()) +
ggplot2::xlab("TFs")
p
The TFs GLI3 and SPDEF are deactivated in KO when
compared to WT, while MUC and NFKB1 seem to be activated.
We can further visualize the most differential target genes in each TF along their
p-values to interpret the results. For example, let's see the genes that are
belong to SP1:
tf <- 'SP1'
df <- net %>%
dplyr::filter(source == tf) %>%
dplyr::arrange(target) %>%
dplyr::mutate(ID = target, color = "3") %>%
tibble::column_to_rownames('target')
inter <- sort(dplyr::intersect(rownames(deg), rownames(df)))
df <- df[inter, ]
df[,c('logfc', 't_value', 'p_value')] <- deg[inter, ]
df <- df %>%
dplyr::mutate(color = dplyr::if_else(mor > 0 & t_value > 0, '1', color)) %>%
dplyr::mutate(color = dplyr::if_else(mor > 0 & t_value < 0, '2', color)) %>%
dplyr::mutate(color = dplyr::if_else(mor < 0 & t_value > 0, '2', color)) %>%
dplyr::mutate(color = dplyr::if_else(mor < 0 & t_value < 0, '1', color))
colors <- rev(RColorBrewer::brewer.pal(n = 11, name = "RdBu")[c(2, 10)])
p <- ggplot2::ggplot(data = df,
mapping = ggplot2::aes(x = logfc,
y = -log10(p_value),
color = color,
size = abs(mor))) +
ggplot2::geom_point(size = 2.5,
color = "black") +
ggplot2::geom_point(size = 1.5) +
ggplot2::scale_colour_manual(values = c(colors[2], colors[1], "grey")) +
ggrepel::geom_label_repel(mapping = ggplot2::aes(label = ID,
size = 1)) +
ggplot2::theme_minimal() +
ggplot2::theme(legend.position = "none") +
ggplot2::geom_vline(xintercept = 0, linetype = 'dotted') +
ggplot2::geom_hline(yintercept = 0, linetype = 'dotted') +
ggplot2::ggtitle(tf)
p
Here blue means that the sign of multiplying the mor and t-value is negative,
meaning that these genes are "deactivating" the TF, and red means that the sign
is positive, meaning that these genes are "activating" the TF.
Session information
options(width = 120)
sessioninfo::session_info()
saezlab/decoupleR documentation built on June 9, 2025, 1:55 p.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
knitr::opts_chunk$set( collapse = TRUE, comment = "#>" )
Bulk RNA-seq yield many molecular readouts that are hard to interpret by themselves. One way of summarizing this information is by inferring transcription factor (TF) activities from prior knowledge.
In this notebook we showcase how to use decoupleR for transcription factor activity
inference with a bulk RNA-seq data-set where the transcription factor FOXA2 was
knocked out in pancreatic cancer cell lines.
The data consists of 3 Wild Type (WT) samples and 3 Knock Outs (KO). They are freely available in GEO.
Loading packages
First, we need to load the relevant packages:
## We load the required packages library(decoupleR) library(dplyr) library(tibble) library(tidyr) library(ggplot2) library(pheatmap) library(ggrepel)
Loading the data-set
Here we used an already processed bulk RNA-seq data-set. We provide the
normalized log-transformed counts, the experimental design meta-data and the
Differential Expressed Genes (DEGs) obtained using limma.
For this example we use limma but we could have used DeSeq2, edgeR or any
other statistical framework. decoupleR requires a gene level statistic to
perform enrichment analysis but it is agnostic of how it was generated. However,
we do recommend to use statistics that include the direction of change and its
significance, for example the t-value obtained for limma(t) or DeSeq2(stat).
edgeR does not return such statistic but we can create our own by weighting the
obtained logFC by pvalue with this formula: -log10(pvalue) * logFC.
We can open the data like this:
inputs_dir <- system.file("extdata", package = "decoupleR") data <- readRDS(file.path(inputs_dir, "bk_data.rds"))
From data we can extract the mentioned information. Here we see the normalized
log-transformed counts:
# Remove NAs and set row names counts <- data$counts %>% dplyr::mutate_if(~ any(is.na(.x)), ~ dplyr::if_else(is.na(.x), 0, .x)) %>% tibble::column_to_rownames(var = "gene") %>% as.matrix() head(counts)
The design meta-data:
design <- data$design design
And the results of limma, of which we are interested in extracting the
obtained t-value and p-value from the contrast:
# Extract t-values per gene deg <- data$limma_ttop %>% dplyr::select(ID, logFC, t, P.Value) %>% dplyr::filter(!is.na(t)) %>% tibble::column_to_rownames(var = "ID") %>% as.matrix() head(deg)
CollecTRI network
CollecTRI is a comprehensive resource containing a curated collection of TFs and their transcriptional targets compiled from 12 different resources. This collection provides an increased coverage of transcription factors and a superior performance in identifying perturbed TFs compared to our previous DoRothEA network and other literature based GRNs. Similar to DoRothEA, interactions are weighted by their mode of regulation (activation or inhibition).
For this example we will use the human version (mouse and rat are also
available). We can use decoupleR to retrieve it from OmniPath. The argument
split_complexes keeps complexes or splits them into subunits, by default we
recommend to keep complexes together.
net <- decoupleR::get_collectri(organism = 'human', split_complexes = FALSE) net
Activity inference with Univariate Linear Model (ULM)
To infer TF enrichment scores we will run the Univariate Linear Model (ulm) method. For each sample in our dataset (mat) and each TF in our network (net), it fits a linear model that predicts the observed gene expression
based solely on the TF's TF-Gene interaction weights. Once fitted, the obtained t-value of the slope is the score. If it is positive, we interpret that the TF is active and if it is negative we interpret that it is inactive.
To run decoupleR methods, we need an input matrix (mat), an input prior
knowledge network/resource (net), and the name of the columns of net that we
want to use.
# Run ulm sample_acts <- decoupleR::run_ulm(mat = counts, net = net, .source = 'source', .target = 'target', .mor = 'mor', minsize = 5) sample_acts
Visualization
From the obtained results we will observe the most variable activities across samples in a heat-map:
n_tfs <- 25 # Transform to wide matrix sample_acts_mat <- sample_acts %>% tidyr::pivot_wider(id_cols = 'condition', names_from = 'source', values_from = 'score') %>% tibble::column_to_rownames('condition') %>% as.matrix() # Get top tfs with more variable means across clusters tfs <- sample_acts %>% dplyr::group_by(source) %>% dplyr::summarise(std = stats::sd(score)) %>% dplyr::arrange(-abs(std)) %>% head(n_tfs) %>% dplyr::pull(source) sample_acts_mat <- sample_acts_mat[,tfs] # Scale per sample sample_acts_mat <- scale(sample_acts_mat) # Choose color palette colors <- rev(RColorBrewer::brewer.pal(n = 11, name = "RdBu")) colors.use <- grDevices::colorRampPalette(colors = colors)(100) my_breaks <- c(seq(-2, 0, length.out = ceiling(100 / 2) + 1), seq(0.05, 2, length.out = floor(100 / 2))) # Plot pheatmap::pheatmap(mat = sample_acts_mat, color = colors.use, border_color = "white", breaks = my_breaks, cellwidth = 15, cellheight = 15, treeheight_row = 20, treeheight_col = 20)
We can also infer TF activities from the t-values of the DEGs between KO and WT:
# Run ulm contrast_acts <- decoupleR::run_ulm(mat = deg[, 't', drop = FALSE], net = net, .source = 'source', .target = 'target', .mor='mor', minsize = 5) contrast_acts
Let's show the changes in activity between KO and WT:
# Filter top TFs in both signs f_contrast_acts <- contrast_acts %>% dplyr::mutate(rnk = NA) msk <- f_contrast_acts$score > 0 f_contrast_acts[msk, 'rnk'] <- rank(-f_contrast_acts[msk, 'score']) f_contrast_acts[!msk, 'rnk'] <- rank(-abs(f_contrast_acts[!msk, 'score'])) tfs <- f_contrast_acts %>% dplyr::arrange(rnk) %>% head(n_tfs) %>% dplyr::pull(source) f_contrast_acts <- f_contrast_acts %>% filter(source %in% tfs) colors <- rev(RColorBrewer::brewer.pal(n = 11, name = "RdBu")[c(2, 10)]) p <- ggplot2::ggplot(data = f_contrast_acts, mapping = ggplot2::aes(x = stats::reorder(source, score), y = score)) + ggplot2::geom_bar(mapping = ggplot2::aes(fill = score), color = "black", stat = "identity") + ggplot2::scale_fill_gradient2(low = colors[1], mid = "whitesmoke", high = colors[2], midpoint = 0) + ggplot2::theme_minimal() + ggplot2::theme(axis.title = element_text(face = "bold", size = 12), axis.text.x = ggplot2::element_text(angle = 45, hjust = 1, size = 10, face = "bold"), axis.text.y = ggplot2::element_text(size = 10, face = "bold"), panel.grid.major = element_blank(), panel.grid.minor = element_blank()) + ggplot2::xlab("TFs") p
The TFs GLI3 and SPDEF are deactivated in KO when compared to WT, while MUC and NFKB1 seem to be activated.
We can further visualize the most differential target genes in each TF along their p-values to interpret the results. For example, let's see the genes that are belong to SP1:
tf <- 'SP1' df <- net %>% dplyr::filter(source == tf) %>% dplyr::arrange(target) %>% dplyr::mutate(ID = target, color = "3") %>% tibble::column_to_rownames('target') inter <- sort(dplyr::intersect(rownames(deg), rownames(df))) df <- df[inter, ] df[,c('logfc', 't_value', 'p_value')] <- deg[inter, ] df <- df %>% dplyr::mutate(color = dplyr::if_else(mor > 0 & t_value > 0, '1', color)) %>% dplyr::mutate(color = dplyr::if_else(mor > 0 & t_value < 0, '2', color)) %>% dplyr::mutate(color = dplyr::if_else(mor < 0 & t_value > 0, '2', color)) %>% dplyr::mutate(color = dplyr::if_else(mor < 0 & t_value < 0, '1', color)) colors <- rev(RColorBrewer::brewer.pal(n = 11, name = "RdBu")[c(2, 10)]) p <- ggplot2::ggplot(data = df, mapping = ggplot2::aes(x = logfc, y = -log10(p_value), color = color, size = abs(mor))) + ggplot2::geom_point(size = 2.5, color = "black") + ggplot2::geom_point(size = 1.5) + ggplot2::scale_colour_manual(values = c(colors[2], colors[1], "grey")) + ggrepel::geom_label_repel(mapping = ggplot2::aes(label = ID, size = 1)) + ggplot2::theme_minimal() + ggplot2::theme(legend.position = "none") + ggplot2::geom_vline(xintercept = 0, linetype = 'dotted') + ggplot2::geom_hline(yintercept = 0, linetype = 'dotted') + ggplot2::ggtitle(tf) p
Here blue means that the sign of multiplying the mor and t-value is negative,
meaning that these genes are "deactivating" the TF, and red means that the sign
is positive, meaning that these genes are "activating" the TF.
Session information
options(width = 120) sessioninfo::session_info()
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.