In nf-osi/nfportalutils: NF Portal Utilities
knitr::opts_chunk$set(
collapse = TRUE,
comment = "#>"
)
Intro
Purpose
This vignette documents in-practice usage of the annotation utils for nf-processed data files.
The outputs are:
1. A metadata manifest for a processed dataset that can be further validated with schematic before submission.
2. Provenance meta.
Typically, these are inspected/validated before submitting to Synapse as a final followup step.
Examples can be run with READ access only to processed outputs,
but requires you to have DOWNLOAD access or a local copy of the input samplesheet.
To actually apply any annotations of course requires EDIT access.
General idea
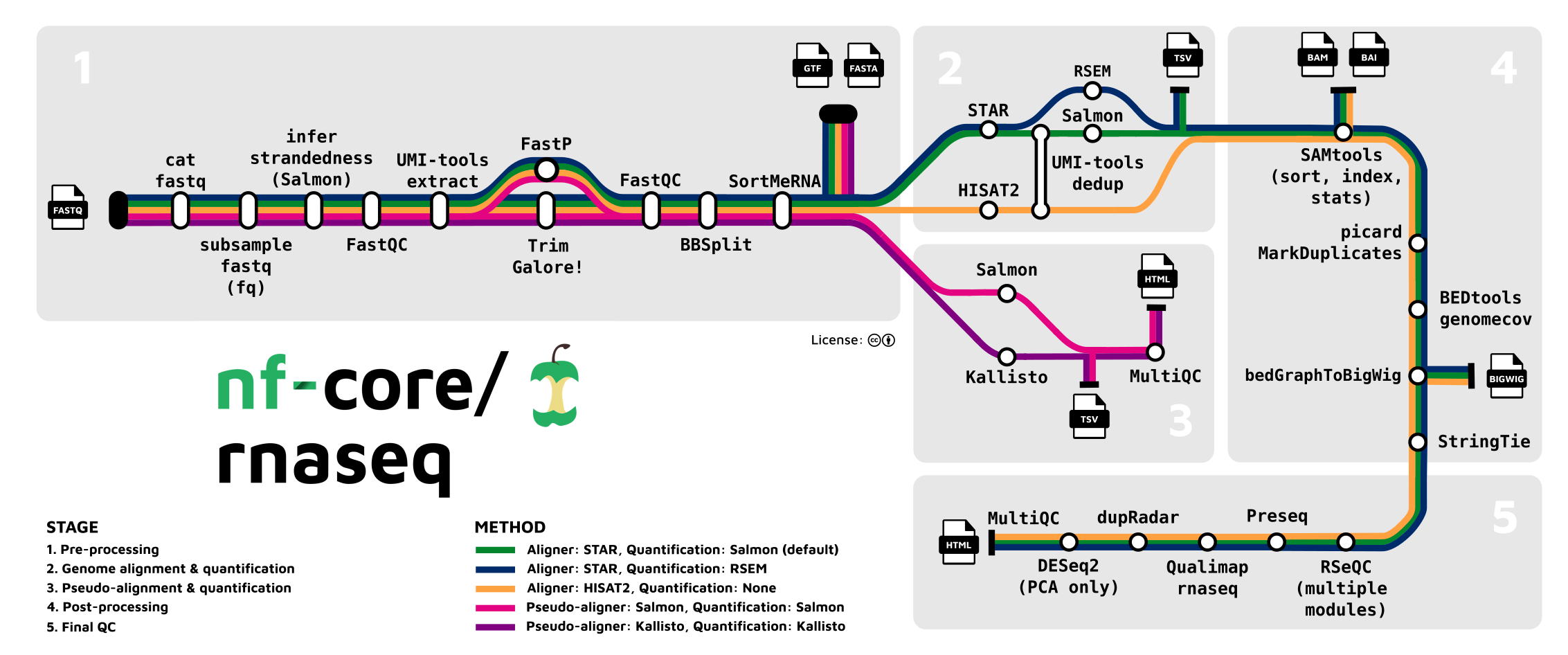
A nextflow workflow generates different types of outputs along the steps in the workflow (see figure above).
At some of these steps/stops, we have products that can be collected and packaged into "level 2, 3, or 4" datasets.
For example, the .bam/bai outputs from SAMtools represent a "level 2" semi-processed dataset with a dataType of AlignedReads.
Ideally, we would simply like to point to the main folder containing all processed output files and
get back a list of manifests that represent all useful dataset products from the workflow.
(What is a "useful", selectable data product is encoded in this annotation workflow.)
These manifests can then be used to annotate the files as well as for creation of Synapse Datasets.
Set up
First load the nfportalutils package and log in.
The recommended default usage of syn_login is to use it without directly passing in credentials.
Instead, have available the SYNAPSE_AUTH_TOKEN environment variable with your token stored therein.
library(nfportalutils)
library(data.table)
syn_login()
Steps
The general annotation workflow steps are:
1. Parse the input samplesheet.
2. Get basic context of processed outputs from the workflow run.
Because none of the indexed-back output files have annotations,
we have to first construct initial info sample, caller, etc. out of the good ol' folder hierarchy and file names.
3. Now with some sample and workflow context at least, link input-output data appropriately, check sample correspondence,
and get into format expected for downstream.
4. Transfer other meta from input to output processed files (most important are individualID, basic individual attributes, assay).
5. Set annotations for processed data type based on workflow default rules.
Some potential issues should be noted:
- If input files have missing or incorrect annotations, processed files will have missing or incorrect annotations.
- If sample ids and other information are updated on the original raw input files, data must be reannotated by rerunning the pipeline.
- Anything that deviates from a relatively standard workflow run, leading to changes in locations or naming of outputs,
might yield poor results or require more manual composition of steps. Standard organization and naming of files is very important.
nf-rnaseq
What does output look like?
Use ?map_sample_output_rnaseq to see which outputs are handled in the parameter output.
But note that depending on how the workflow was run and data indexed back into Synapse, actual output availability may differ.
In some projects, bam/bai files may not even be indexed back into Synapse.
As an illustrative example, the workflow outputs here does not include featureCounts:
syn_out <- "syn57382909"
fileview <- "syn11601495"
o <- map_sample_output_rnaseq(syn_out, fileview) # check outputs only
names(o)
What does input look like?
Like output, input is just another index of files and is actually the samplesheet used by the workflow to know what files to process. Samplesheets should be public and placed in the pipeline_info directory as part of the workflow (most of the time).
IMPORTANT: The samplesheet needs to be standard enough to parse correctly, i.e. to extract valid file Synapse ids from the first fastq. We use the same helper to parse samplesheets for the two workflows (both RNA-seq and Sarek), and the function will do its best to handle slight variations in samplesheet formats. Here are examples of what will work vs not:
- ✔ OK. Excerpt from real samplesheet syn51525432.
ss1 <- data.frame(
sample = c("JH-2-019-DB5EH-C461C", "JH-2-007-B14BB-AG2A6", "JH-2-009-518B9-77BH3"),
fastq_1 = c("syn15261791", "syn15261974", "syn15262157"),
fastq_2 = c("syn15261900", "syn15262033", "syn15262216"),
strandedness = c("auto", "auto", "auto"),
stringsAsFactors = FALSE
)
ss1
- ✔ OK. Excerpt from real samplesheet syn63172939.
ss2 <- data.frame(
subject = c("JHU002", "JHU002", "JHU023"),
sex = c("XY", "XY", "XY"),
status = c(1, 1, 1),
sample = c("JHU002-043", "JHU002-048", "JHU023-044"),
lane = c("JHU002-043-Lane-1", "JHU002-048-Lane-1", "JHU023-044-Lane-1"),
fastq1 = c("syn://syn22091879", "syn://syn22091925", "syn://syn22091973"),
fastq2 = c(NA, NA, NA),
datasetId = c("syn29783617", "syn29783617", "syn29783617"),
projectId = c("syn11638893", "syn11638893", "syn11638893"),
output_destination_id = c("syn29429576", "syn29429576", "syn29429576"),
Germline = c("Y", "Y", "Y"),
Somatic = c(NA, NA, NA),
stringsAsFactors = FALSE
)
ss2
- ✖ No. Adapted from real samplesheet syn63172939.
This will give an error because "x6" is not a valid Synapse ID. A manually corrected samplesheet will have to be provided.
ss3 <- data.frame(
sample = c("patient10tumor1_T1", "patient10tumor2_T1", "patient10tumor3_T1"),
single_end = c(0, 0, 0),
fastq_1 = c(
"s3://some-tower-bucket/syn40134517/x6/SL106309_1.fastq.gz",
"s3://some-tower-bucket/syn40134517/syn7989846/SL106310_1.fastq.gz",
"s3://some-tower-bucket/syn40134517/syn7989852/SL106311_1.fastq.gz"
),
fastq_2 = c(
"s3://some-tower-bucket/syn40134517/syn7989839/SL106309_2.fastq.gz",
"s3://some-tower-bucket/syn40134517/syn7989847/SL106310_2.fastq.gz",
"s3://some-tower-bucket/syn40134517/syn7989856/SL106311_2.fastq.gz"
),
strandedness = c("auto", "auto", "auto"),
stringsAsFactors = FALSE
)
ss3
Connecting input and output to automate filled manifests
In contrast with the previous example, run this other example for an output directory that does all types of outputs we're looking for in an nf-rnaseq workflow. This will be used for the rest of the demo.
(Review the source code for processed_meta to see the steps encapsulated.)
samplesheet <- "syn51408030"
syn_out <- "syn51476810"
fileview <- "syn11601481"
wf_link <- "https://nf-co.re/rnaseq/3.11.2/output#star-and-salmon"
input <- map_sample_input_ss(samplesheet)
# Alternatively, use a local file if not on Synapse:
# input <- map_sample_input_ss("~/work/samplesheet.csv")
output <- map_sample_output_rnaseq(syn_out, fileview)
names(output)
Generate the manifests and inspect an example result:
meta <- processed_meta(input, output, workflow_link = wf_link)
head(meta$manifests$SAMtools)
Submit manifest
Manifests can be submitted with schematic-compatible or using annotate_with_manifest as shown below.
mannifest_1 <- meta$manifests$SAMtools
annotate_with_manifest(manifest_1)
Add provenance
Provenance is basically an annotation, though treated somewhat differently in Synapse.
In the result meta object, there is something called sample_io that can be provided to add_activity_batch to add provenance.
"Workflow" provides the general name to the activity,
while "workflow link" provides a more persistent reference to some version/part of the workflow,
which others can follow the link to get details.
sample_io <- meta$sample_io
prov <- add_activity_batch(sample_io$output_id,
sample_io$workflow,
wf_link,
sample_io$input_id)
Create dataset
To create a Synapse Dataset:
items <- manifest_1$entityId
project <- "your-dev-project-synid"
dataset_1 <- new_dataset(name = "STAR Salmon Gene Expression Quantification from RNA-seq",
parent = project,
items = items,
dry_run = FALSE)
nf-sarek
The only difference is usage of map_sample_output_sarek at Step 2.
samplesheet <- "syn38793905" # samplesheet can be stored on Synapse or locally
syn_out <- "syn27650634"
fileview <- "syn13363852"
i <- map_sample_input_ss(samplesheet) #1
o <- map_sample_output_sarek(syn_out, fileview) #2
sarek_meta <- processed_meta(i, o, workflow_link = "test")
# View first manifest
sarek_meta$manifests$Strelka2
Add provenance
Use the manifest to add provenance.
wf_link <- c(FreeBayes = "https://nf-co.re/sarek/3.2.3/output#freebayes",
Mutect2 = "https://nf-co.re/sarek/3.2.3/output#gatk-mutect2",
Strelka2 = "https://nf-co.re/sarek/3.2.3/output#strelka2")
sample_io <- sarek_meta$sample_io
add_activity_batch(sample_io$output_id,
sample_io$workflow,
wf_link[sample_io$workflow],
sample_io$input_id)
After provenance, the rest of the workflow for manifest submission or creating datasets is like the nf-rnaseq example.
nf-osi/nfportalutils documentation built on June 10, 2025, 5:08 a.m.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
knitr::opts_chunk$set( collapse = TRUE, comment = "#>" )
Intro
Purpose
This vignette documents in-practice usage of the annotation utils for nf-processed data files.
The outputs are:
1. A metadata manifest for a processed dataset that can be further validated with schematic before submission.
2. Provenance meta.
Typically, these are inspected/validated before submitting to Synapse as a final followup step.
Examples can be run with READ access only to processed outputs, but requires you to have DOWNLOAD access or a local copy of the input samplesheet. To actually apply any annotations of course requires EDIT access.
General idea
A nextflow workflow generates different types of outputs along the steps in the workflow (see figure above).
At some of these steps/stops, we have products that can be collected and packaged into "level 2, 3, or 4" datasets.
For example, the .bam/bai outputs from SAMtools represent a "level 2" semi-processed dataset with a dataType of AlignedReads.
Ideally, we would simply like to point to the main folder containing all processed output files and get back a list of manifests that represent all useful dataset products from the workflow. (What is a "useful", selectable data product is encoded in this annotation workflow.)
These manifests can then be used to annotate the files as well as for creation of Synapse Datasets.
Set up
First load the nfportalutils package and log in.
The recommended default usage of syn_login is to use it without directly passing in credentials.
Instead, have available the SYNAPSE_AUTH_TOKEN environment variable with your token stored therein.
library(nfportalutils) library(data.table) syn_login()
Steps
The general annotation workflow steps are:
1. Parse the input samplesheet.
2. Get basic context of processed outputs from the workflow run.
Because none of the indexed-back output files have annotations,
we have to first construct initial info sample, caller, etc. out of the good ol' folder hierarchy and file names.
3. Now with some sample and workflow context at least, link input-output data appropriately, check sample correspondence,
and get into format expected for downstream.
4. Transfer other meta from input to output processed files (most important are individualID, basic individual attributes, assay).
5. Set annotations for processed data type based on workflow default rules.
Some potential issues should be noted:
- If input files have missing or incorrect annotations, processed files will have missing or incorrect annotations.
- If sample ids and other information are updated on the original raw input files, data must be reannotated by rerunning the pipeline.
- Anything that deviates from a relatively standard workflow run, leading to changes in locations or naming of outputs, might yield poor results or require more manual composition of steps. Standard organization and naming of files is very important.
nf-rnaseq
What does output look like?
Use ?map_sample_output_rnaseq to see which outputs are handled in the parameter output.
But note that depending on how the workflow was run and data indexed back into Synapse, actual output availability may differ.
In some projects, bam/bai files may not even be indexed back into Synapse. As an illustrative example, the workflow outputs here does not include featureCounts:
syn_out <- "syn57382909" fileview <- "syn11601495" o <- map_sample_output_rnaseq(syn_out, fileview) # check outputs only names(o)
What does input look like?
Like output, input is just another index of files and is actually the samplesheet used by the workflow to know what files to process. Samplesheets should be public and placed in the pipeline_info directory as part of the workflow (most of the time).
IMPORTANT: The samplesheet needs to be standard enough to parse correctly, i.e. to extract valid file Synapse ids from the first fastq. We use the same helper to parse samplesheets for the two workflows (both RNA-seq and Sarek), and the function will do its best to handle slight variations in samplesheet formats. Here are examples of what will work vs not:
- ✔ OK. Excerpt from real samplesheet syn51525432.
ss1 <- data.frame( sample = c("JH-2-019-DB5EH-C461C", "JH-2-007-B14BB-AG2A6", "JH-2-009-518B9-77BH3"), fastq_1 = c("syn15261791", "syn15261974", "syn15262157"), fastq_2 = c("syn15261900", "syn15262033", "syn15262216"), strandedness = c("auto", "auto", "auto"), stringsAsFactors = FALSE ) ss1
- ✔ OK. Excerpt from real samplesheet syn63172939.
ss2 <- data.frame( subject = c("JHU002", "JHU002", "JHU023"), sex = c("XY", "XY", "XY"), status = c(1, 1, 1), sample = c("JHU002-043", "JHU002-048", "JHU023-044"), lane = c("JHU002-043-Lane-1", "JHU002-048-Lane-1", "JHU023-044-Lane-1"), fastq1 = c("syn://syn22091879", "syn://syn22091925", "syn://syn22091973"), fastq2 = c(NA, NA, NA), datasetId = c("syn29783617", "syn29783617", "syn29783617"), projectId = c("syn11638893", "syn11638893", "syn11638893"), output_destination_id = c("syn29429576", "syn29429576", "syn29429576"), Germline = c("Y", "Y", "Y"), Somatic = c(NA, NA, NA), stringsAsFactors = FALSE ) ss2
- ✖ No. Adapted from real samplesheet syn63172939. This will give an error because "x6" is not a valid Synapse ID. A manually corrected samplesheet will have to be provided.
ss3 <- data.frame( sample = c("patient10tumor1_T1", "patient10tumor2_T1", "patient10tumor3_T1"), single_end = c(0, 0, 0), fastq_1 = c( "s3://some-tower-bucket/syn40134517/x6/SL106309_1.fastq.gz", "s3://some-tower-bucket/syn40134517/syn7989846/SL106310_1.fastq.gz", "s3://some-tower-bucket/syn40134517/syn7989852/SL106311_1.fastq.gz" ), fastq_2 = c( "s3://some-tower-bucket/syn40134517/syn7989839/SL106309_2.fastq.gz", "s3://some-tower-bucket/syn40134517/syn7989847/SL106310_2.fastq.gz", "s3://some-tower-bucket/syn40134517/syn7989856/SL106311_2.fastq.gz" ), strandedness = c("auto", "auto", "auto"), stringsAsFactors = FALSE ) ss3
Connecting input and output to automate filled manifests
In contrast with the previous example, run this other example for an output directory that does all types of outputs we're looking for in an nf-rnaseq workflow. This will be used for the rest of the demo.
(Review the source code for processed_meta to see the steps encapsulated.)
samplesheet <- "syn51408030" syn_out <- "syn51476810" fileview <- "syn11601481" wf_link <- "https://nf-co.re/rnaseq/3.11.2/output#star-and-salmon" input <- map_sample_input_ss(samplesheet) # Alternatively, use a local file if not on Synapse: # input <- map_sample_input_ss("~/work/samplesheet.csv") output <- map_sample_output_rnaseq(syn_out, fileview) names(output)
Generate the manifests and inspect an example result:
meta <- processed_meta(input, output, workflow_link = wf_link) head(meta$manifests$SAMtools)
Submit manifest
Manifests can be submitted with schematic-compatible or using annotate_with_manifest as shown below.
mannifest_1 <- meta$manifests$SAMtools annotate_with_manifest(manifest_1)
Add provenance
Provenance is basically an annotation, though treated somewhat differently in Synapse.
In the result meta object, there is something called sample_io that can be provided to add_activity_batch to add provenance.
"Workflow" provides the general name to the activity, while "workflow link" provides a more persistent reference to some version/part of the workflow, which others can follow the link to get details.
sample_io <- meta$sample_io prov <- add_activity_batch(sample_io$output_id, sample_io$workflow, wf_link, sample_io$input_id)
Create dataset
To create a Synapse Dataset:
items <- manifest_1$entityId project <- "your-dev-project-synid" dataset_1 <- new_dataset(name = "STAR Salmon Gene Expression Quantification from RNA-seq", parent = project, items = items, dry_run = FALSE)
nf-sarek
The only difference is usage of map_sample_output_sarek at Step 2.
samplesheet <- "syn38793905" # samplesheet can be stored on Synapse or locally syn_out <- "syn27650634" fileview <- "syn13363852" i <- map_sample_input_ss(samplesheet) #1 o <- map_sample_output_sarek(syn_out, fileview) #2 sarek_meta <- processed_meta(i, o, workflow_link = "test") # View first manifest sarek_meta$manifests$Strelka2
Add provenance
Use the manifest to add provenance.
wf_link <- c(FreeBayes = "https://nf-co.re/sarek/3.2.3/output#freebayes", Mutect2 = "https://nf-co.re/sarek/3.2.3/output#gatk-mutect2", Strelka2 = "https://nf-co.re/sarek/3.2.3/output#strelka2") sample_io <- sarek_meta$sample_io add_activity_batch(sample_io$output_id, sample_io$workflow, wf_link[sample_io$workflow], sample_io$input_id)
After provenance, the rest of the workflow for manifest submission or creating datasets is like the nf-rnaseq example.
R Package Documentation
Browse R Packages
We want your feedback!
Note that we can't provide technical support on individual packages. You should contact the package authors for that.
Embedding an R snippet on your website
Add the following code to your website.
For more information on customizing the embed code, read Embedding Snippets.